

Description
Simpson-Golabi-Behmel syndrome is a condition that affects many parts of the body and occurs primarily in males. This condition is classified as an overgrowth syndrome, which means that affected infants are considerably larger than normal at birth (macrosomia) and continue to grow and gain weight at an unusual rate. The other signs and symptoms of Simpson-Golabi-Behmel syndrome vary widely. People with mild cases often live into adulthood.
People with Simpson-Golabi-Behmel syndrome have distinctive facial features including widely spaced eyes (ocular hypertelorism), an unusually large mouth (macrostomia), a large tongue (macroglossia) that may have a deep groove or furrow down the middle, a broad nose with an upturned tip, and abnormalities affecting the roof of the mouth (the palate). The facial features are often described as "coarse" in older children and adults with this condition.



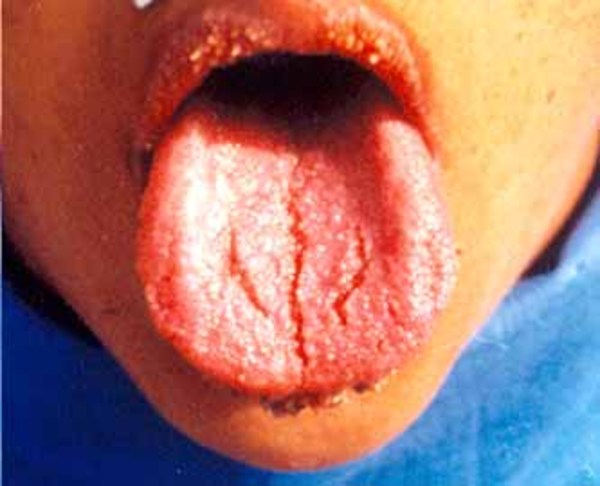
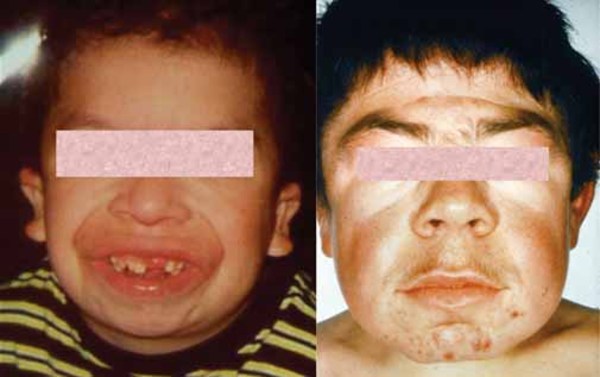
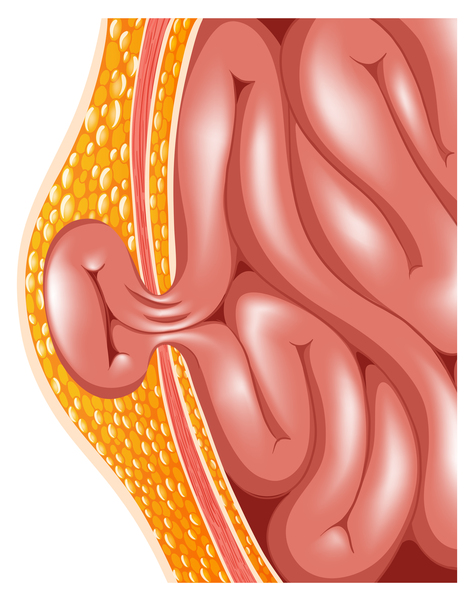
Other features of Simpson-Golabi-Behmel syndrome involve the chest and abdomen. Affected infants may be born with one or more extra nipples, an abnormal opening in the muscle covering the abdomen (diastasis recti), a soft out-pouching around the belly-button (an umbilical hernia), or a hole in the diaphragm (a diaphragmatic hernia) that allows the stomach and intestines to move into the chest and crowd the developing heart and lungs.

Simpson-Golabi-Behmel syndrome can also cause heart defects, malformed or abnormally large kidneys, an enlarged liver and spleen (hepatosplenomegaly), and skeletal abnormalities. Additionally, the syndrome can affect the development of the gastrointestinal system, urinary system, and genitalia. Some people with this condition have mild to severe intellectual disability, while others have normal intelligence.

About 10 percent of people with Simpson-Golabi-Behmel syndrome develop cancerous or noncancerous tumors in early childhood. The most common tumors are a rare form of kidney cancer called Wilms tumor and a cancerous tumor called a neuroblastoma that arises from developing nerve cells.
Frequency
The incidence of Simpson-Golabi-Behmel syndrome is unknown. At least 250 people worldwide have been diagnosed with this disorder.
Causes
Mutations in the GPC3 gene are the most common cause of Simpson-Golabi-Behmel syndrome. This gene provides instructions for making a protein called glypican 3, which blocks (inhibits) a developmental pathway called the hedgehog signaling pathway. This pathway is critical for cell growth and division (proliferation), cell specialization, and the normal shaping (patterning) of many parts of the body during embryonic development. Researchers believe that glypican 3 also helps establish the body's shape by causing certain cells to self-destruct (undergo apoptosis) when they are no longer needed.
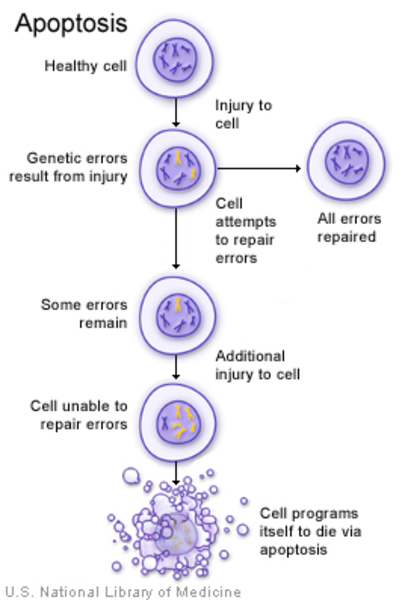
GPC3 gene mutations prevent glypican 3 from inhibiting the hedgehog signaling pathway. The resulting overactivity of this pathway leads to an increased rate of cell growth and division starting before birth. This increased cell proliferation accounts, at least in part, for the overgrowth that occurs in Simpson-Golabi-Behmel syndrome. It is unclear how changes in hedgehog signaling contribute to the other abnormalities that can occur with this disorder.
Some individuals with Simpson-Golabi-Behmel syndrome do not have an identified mutation in the GPC3 gene. Mutations in other genes have been studied as possible causes of this condition, but most of these genetic changes have been described only in single families or have not been confirmed in subsequent studies. In some cases, the cause of the condition is unknown.
Researchers have described a disorder with features overlapping those of Simpson-Golabi-Behmel syndrome, which they designated as Simpson-Golabi-Behmel syndrome type 2 (SGBS2). The signs and symptoms of this disorder are more severe than those that typically occur with Simpson-Golabi-Behmel syndrome, and affected individuals live only into infancy. This more severe disorder likely has a different genetic cause. Because of these differences, many researchers now consider SGBS2 to be a separate condition, distinct from Simpson-Golabi-Behmel syndrome.
Inheritance
This condition is inherited in an X-linked pattern. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes in each cell. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. Because females have two copies of the X chromosome, one altered copy of the gene in each cell usually leads to less severe health problems in females than in males, or it may cause no signs or symptoms at all.

Some females who have one altered copy of the GPC3 gene have distinctive facial features including an upturned nose, a wide mouth, and a prominent chin. Their fingernails may be malformed and they can have extra nipples. Skeletal abnormalities, including extra spinal bones (vertebrae), are also possible in affected females. Other females who carry one altered copy of the GPC3 gene do not have any health problems associated with Simpson-Golabi-Behmel syndrome.
Other Names for This Condition
- DGSX
- Mental retardation-overgrowth syndrome
- SDYS
- SGBS
- SGBS1
- Simpson dysplasia syndrome
- Simpson syndrome
- Simpson-Golabi-Behmel syndrome type 1
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Capurro MI, Xu P, Shi W, Li F, Jia A, Filmus J. Glypican-3 inhibits Hedgehog signaling during development by competing with patched for Hedgehog binding. Dev Cell. 2008 May;14(5):700-11. doi: 10.1016/j.devcel.2008.03.006. Citation on PubMed
- Cottereau E, Mortemousque I, Moizard MP, Burglen L, Lacombe D, Gilbert-Dussardier B, Sigaudy S, Boute O, David A, Faivre L, Amiel J, Robertson R, Viana Ramos F, Bieth E, Odent S, Demeer B, Mathieu M, Gaillard D, Van Maldergem L, Baujat G, Maystadt I, Heron D, Verloes A, Philip N, Cormier-Daire V, Froute MF, Pinson L, Blanchet P, Sarda P, Willems M, Jacquinet A, Ratbi I, Van Den Ende J, Lackmy-Port Lis M, Goldenberg A, Bonneau D, Rossignol S, Toutain A. Phenotypic spectrum of Simpson-Golabi-Behmel syndrome in a series of 42 cases with a mutation in GPC3 and review of the literature. Am J Med Genet C Semin Med Genet. 2013 May;163C(2):92-105. doi: 10.1002/ajmg.c.31360. Epub 2013 Apr 18. Citation on PubMed
- Fauth C, Steindl K, Toutain A, Farrell S, Witsch-Baumgartner M, Karall D, Joset P, Bohm S, Baumer A, Maier O, Zschocke J, Weksberg R, Marshall CR, Rauch A. A recurrent germline mutation in the PIGA gene causes Simpson-Golabi-Behmel syndrome type 2. Am J Med Genet A. 2016 Feb;170A(2):392-402. doi: 10.1002/ajmg.a.37452. Epub 2015 Nov 6. Citation on PubMed
- Kehrer C, Hoischen A, Menkhaus R, Schwab E, Muller A, Kim S, Kreiss M, Weitensteiner V, Hilger A, Berg C, Geipel A, Reutter H, Gembruch U. Whole exome sequencing and array-based molecular karyotyping as aids to prenatal diagnosis in fetuses with suspected Simpson-Golabi-Behmel syndrome. Prenat Diagn. 2016 Oct;36(10):961-965. doi: 10.1002/pd.4920. Epub 2016 Sep 27. Citation on PubMed
- Nisbet AF, Hathaway ER, Kalish JM. Simpson-Golabi-Behmel Syndrome Type 1. 2006 Dec 19 [updated 2024 Nov 14]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1219/ Citation on PubMed
- Sakazume S, Okamoto N, Yamamoto T, Kurosawa K, Numabe H, Ohashi Y, Kako Y, Nagai T, Ohashi H. GPC3 mutations in seven patients with Simpson-Golabi-Behmel syndrome. Am J Med Genet A. 2007 Aug 1;143A(15):1703-7. doi: 10.1002/ajmg.a.31822. Citation on PubMed
- Tenorio J, Arias P, Martinez-Glez V, Santos F, Garcia-Minaur S, Nevado J, Lapunzina P. Simpson-Golabi-Behmel syndrome types I and II. Orphanet J Rare Dis. 2014 Sep 20;9:138. doi: 10.1186/s13023-014-0138-0. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.