

Description
Wilms tumor is a form of kidney cancer that primarily develops in children. Nearly all cases of Wilms tumor are diagnosed before the age of 10, with two-thirds being found before age 5.
Wilms tumor is often first noticed because of abdominal swelling or a mass in the kidney that can be felt upon physical examination. Some affected children have abdominal pain, fever, a low number of red blood cells (anemia), blood in the urine (hematuria), or high blood pressure (hypertension). Additional signs of Wilms tumor can include loss of appetite, weight loss, nausea, vomiting, and tiredness (lethargy).

Wilms tumor can develop in one or both kidneys. About 5 to 10 percent of affected individuals develop multiple tumors in one or both kidneys. Wilms tumor may spread from the kidneys to other parts of the body (metastasize). In rare cases, Wilms tumor does not involve the kidneys and occurs instead in the genital tract, bladder, abdomen, chest, or lower back. It is unclear how Wilms tumor develops in these tissues.
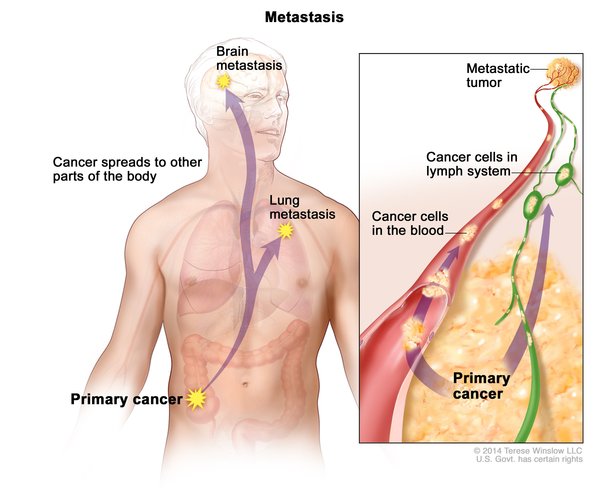

With proper treatment, children with Wilms tumor have a 90 percent survival rate. However, the risk that the cancer will come back (recur) is between 15 and 50 percent, depending on traits of the original tumor. Tumors usually recur in the first 2 years following treatment and develop in the kidneys or other tissues, such as the lungs. Individuals who have had Wilms tumor may experience related health problems or late effects of their treatment in adulthood, such as decreased kidney function, heart disease, and development of additional cancers.
Frequency
Wilms tumor is the most common kidney cancer in children. In Europe and North America, Wilms tumor affects 1 in 10,000 children. In the United States, 500 children develop Wilms tumor each year. The incidence of Wilms tumor seems to vary among populations, with African Americans having a higher-than-average risk of developing this cancer and Asians having a lower-than-average risk.
Wilms tumor rarely develops in adults; only about 300 such cases have been described.
Causes
Changes in any of several genes are involved in the formation of Wilms tumor. Wilms tumor is often associated with variants (also called mutations) in the WT1 gene, CTNNB1 gene, or AMER1 gene. These genes provide instructions for making proteins that regulate gene activity and promote the growth and division (proliferation) of cells. WT1, CTNNB1, and AMER1 gene variants all lead to the unchecked proliferation of cells, allowing tumor development.
Changes on the short (p) arm of chromosome 11 are also associated with developing Wilms tumor. Two genes in this area, IGF2 and H19, are either turned on or off depending on whether the copy of the gene was inherited from the mother or the father. This parent-specific difference in gene activation is a phenomenon called genomic imprinting. In some cases of Wilms tumor, abnormalities in the process of genomic imprinting on chromosome 11 lead to a loss of H19 gene activity and increased activity of the IGF2 gene in kidney cells. The resulting loss of H19 gene activity, which normally restrains cell growth, and increase in IGF2 gene activity, which promotes cell growth, together lead to uncontrolled cell growth and tumor development in people with Wilms tumor.
In most cases of Wilms tumors involving one kidney and nearly all cases involving both kidneys, the tumors are thought to arise from immature kidney tissue that never developed properly. These immature tissues are known as nephrogenic rests. It is likely that genetic changes are involved in the presence of nephrogenic rests and that additional genetic changes trigger nephrogenic rests to develop into a tumor.
Genetic conditions that share a genetic cause with Wilms tumor can also have this cancer as a feature. These conditions include WAGR syndrome, Denys-Drash syndrome, and Frasier syndrome, which are caused by variants in the WT1 gene. Wilms tumor has also been seen in individuals with Beckwith-Wiedemann syndrome, which can be caused by changes in the genomic imprinting of the IGF2 and H19 genes. Wilms tumor can be a feature of other genetic conditions caused by variants in other genes.
Many children with Wilms tumor do not have identified variants in any of the known genes. In these cases, the cause of the condition is unknown. It is likely that other, unknown genes are also associated with the development of Wilms tumor.
Inheritance
Most cases of Wilms tumor are not caused by inherited genetic factors and do not cluster in families. Approximately 90 percent of these cancers are due to somatic variants, which means that the variants are acquired during a person's lifetime and are present only in the tumor cells.
Variants that are present in cells throughout the body (called germline variants) are responsible for the remaining 10 percent of Wilms tumor cases and cause either Wilms tumor without any other signs or symptoms or syndromes in which Wilms tumor is one of multiple features. These cases follow autosomal dominant inheritance , which means one copy of the altered gene in each cell can cause a Wilms tumor-related syndrome or increase a person's chance of developing the cancer alone. Most of these cases result from new (de novo) variants in the gene that occur during the formation of reproductive cells (eggs or sperm) or in early embryonic development.
, which means one copy of the altered gene in each cell can cause a Wilms tumor-related syndrome or increase a person's chance of developing the cancer alone. Most of these cases result from new (de novo) variants in the gene that occur during the formation of reproductive cells (eggs or sperm) or in early embryonic development.
The AMER1 gene is located on the X chromosome (one of the two sex chromosomes ), so when Wilms tumor is caused by variants in this gene, the condition follows an X-linked dominant pattern
), so when Wilms tumor is caused by variants in this gene, the condition follows an X-linked dominant pattern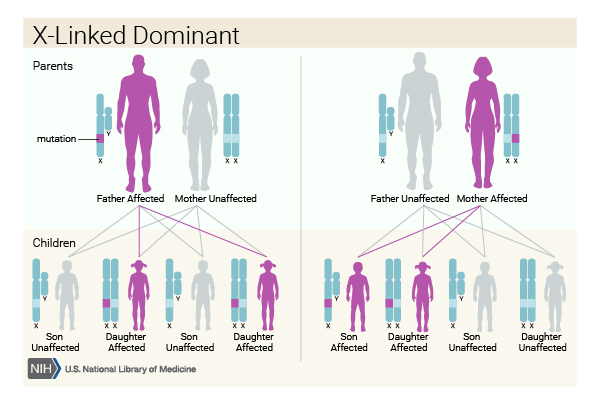 . In females (who have two X chromosomes), a variant in one of the two copies of the gene in each cell is sufficient to increase a person's chance of developing cancer. In males (who have only one X chromosome), a variant in the only copy of the gene in each cell increases their cancer risk.
. In females (who have two X chromosomes), a variant in one of the two copies of the gene in each cell is sufficient to increase a person's chance of developing cancer. In males (who have only one X chromosome), a variant in the only copy of the gene in each cell increases their cancer risk.
In many cases, the genetic basis for Wilms tumor and the mechanism of inheritance are unclear.
Other Names for This Condition
- Embryonal adenosarcoma
- Embryonal nephroma
- Kidney Wilms tumor
- Kidney, adenomyosarcoma, embryonal
- Kidney, carcinosarcoma, embryonal
- Kidney, embryoma
- Kidney, embryonal mixed tumor
- Nephroblastoma
- Nephroma
- Renal adenosarcoma
- Renal cancer, Wilms
- Renal Wilms tumor
- Tumor, Wilms
- Wilms' tumor
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Al-Hussain T, Ali A, Akhtar M. Wilms tumor: an update. Adv Anat Pathol. 2014 May;21(3):166-73. doi: 10.1097/PAP.0000000000000017. Citation on PubMed
- Deng C, Dai R, Li X, Liu F. Genetic variation frequencies in Wilms' tumor: A meta-analysis and systematic review. Cancer Sci. 2016 May;107(5):690-9. doi: 10.1111/cas.12910. Epub 2016 Mar 18. Citation on PubMed or Free article on PubMed Central
- Gadd S, Huff V, Walz AL, Ooms AHAG, Armstrong AE, Gerhard DS, Smith MA, Guidry Auvil JM, Meerzaman D, Chen QR, Hsu CH, Yan C, Nguyen C, Hu Y, Hermida LC, Davidsen T, Gesuwan P, Ma Y, Zong Z, Mungall AJ, Moore RA, Marra MA, Dome JS, Mullighan CG, Ma J, Wheeler DA, Hampton OA, Ross N, Gastier-Foster JM, Arold ST, Perlman EJ. A Children's Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat Genet. 2017 Oct;49(10):1487-1494. doi: 10.1038/ng.3940. Epub 2017 Aug 21. Citation on PubMed or Free article on PubMed Central
- Kalish JM, Doros L, Helman LJ, Hennekam RC, Kuiper RP, Maas SM, Maher ER, Nichols KE, Plon SE, Porter CC, Rednam S, Schultz KAP, States LJ, Tomlinson GE, Zelley K, Druley TE. Surveillance Recommendations for Children with Overgrowth Syndromes and Predisposition to Wilms Tumors and Hepatoblastoma. Clin Cancer Res. 2017 Jul 1;23(13):e115-e122. doi: 10.1158/1078-0432.CCR-17-0710. Citation on PubMed or Free article on PubMed Central
- Salvatorelli L, Parenti R, Leone G, Musumeci G, Vasquez E, Magro G. Wilms tumor 1 (WT1) protein: Diagnostic utility in pediatric tumors. Acta Histochem. 2015 May-Jun;117(4-5):367-78. doi: 10.1016/j.acthis.2015.03.010. Epub 2015 Apr 14. Citation on PubMed
- Tian F, Yourek G, Shi X, Yang Y. The development of Wilms tumor: from WT1 and microRNA to animal models. Biochim Biophys Acta. 2014 Aug;1846(1):180-7. doi: 10.1016/j.bbcan.2014.07.003. Epub 2014 Jul 11. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.