

Description
WAGR syndrome is a disorder that affects many body systems and is named for its main features: Wilms tumor, aniridia, genitourinary anomalies, and a range of developmental delays.
People with WAGR syndrome have a 45 to 60 percent chance of developing Wilms tumor, a rare form of kidney cancer. This type of cancer is most often diagnosed in children but is sometimes seen in adults. Some people with WAGR syndrome develop nephrogenic rests, which are abnormal clumps of cells in the kidneys. These can lead to Wilms tumor, but some people with nephrogenic rests never develop Wilms tumor.
Most people with WAGR syndrome have aniridia, an absence of the colored part of the eye (the iris). This can reduce the sharpness of a person's vision (visual acuity) and increase sensitivity to light (photophobia). Aniridia is typically the first noticeable sign of WAGR syndrome. Other eye problems may also develop, such as clouding of the lens of the eyes (cataracts
(the iris). This can reduce the sharpness of a person's vision (visual acuity) and increase sensitivity to light (photophobia). Aniridia is typically the first noticeable sign of WAGR syndrome. Other eye problems may also develop, such as clouding of the lens of the eyes (cataracts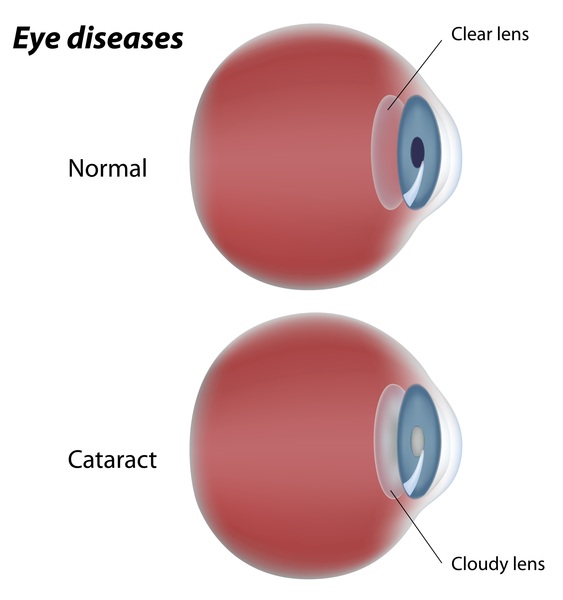 ), increased pressure in the eyes (glaucoma
), increased pressure in the eyes (glaucoma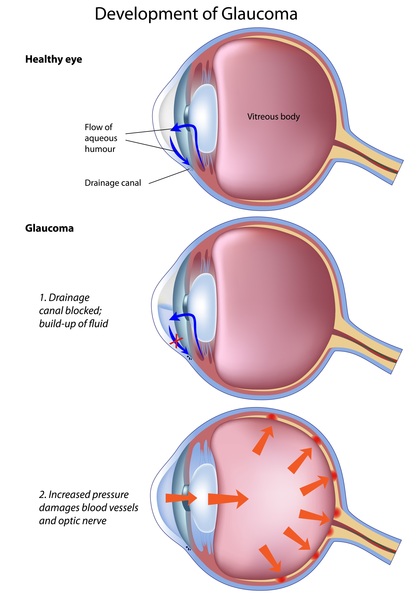 ), and involuntary eye movements (nystagmus).
), and involuntary eye movements (nystagmus).
Abnormalities of the genitalia and urinary tract (genitourinary anomalies) are seen more frequently in males with WAGR syndrome than in affected females. The most common genitourinary abnormality in affected males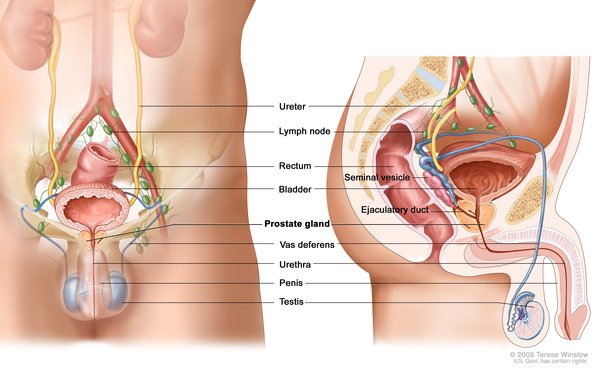 is undescended testes (cryptorchidism). Affected females
is undescended testes (cryptorchidism). Affected females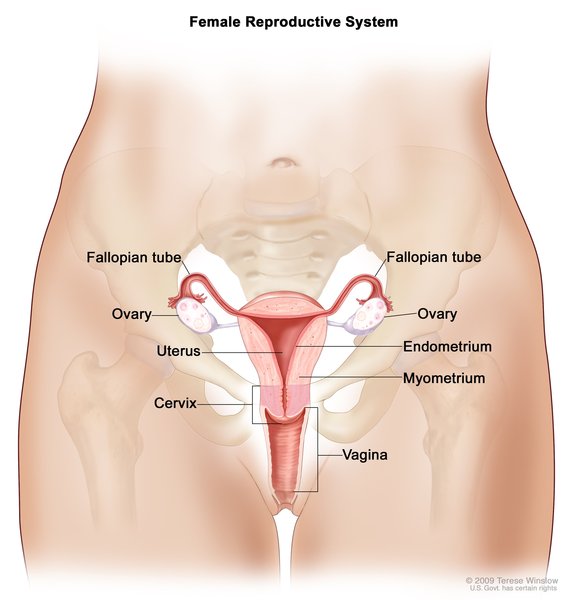 may not have functional ovaries and may instead have undeveloped clumps of tissue called streak gonads. Females with WAGR syndrome may also have a heart-shaped (bicornate) uterus, which makes it difficult to carry a pregnancy to term.
may not have functional ovaries and may instead have undeveloped clumps of tissue called streak gonads. Females with WAGR syndrome may also have a heart-shaped (bicornate) uterus, which makes it difficult to carry a pregnancy to term.
Intellectual disability and other developmental delays are also common in people with WAGR syndrome. Affected individuals often have difficulty processing, learning, and properly responding to information. Many affected individuals have difficulty speaking or understanding language. Some individuals with WAGR syndrome also have psychiatric or behavioral problems such as depression, anxiety, attention-deficit/hyperactivity disorder (ADHD), obsessive-compulsive disorder (OCD), or a developmental disorder called autism spectrum disorder that affects communication and social interaction.
Other signs and symptoms of WAGR syndrome can include ongoing constipation, inflammation of the pancreas (pancreatitis), kidney failure, breathing problems, and allergies. Some affected children have obesity. When WAGR syndrome includes childhood-onset obesity, it is often referred to as WAGRO syndrome.
(pancreatitis), kidney failure, breathing problems, and allergies. Some affected children have obesity. When WAGR syndrome includes childhood-onset obesity, it is often referred to as WAGRO syndrome.
Frequency
The prevalence of WAGR syndrome ranges from 1 in 500,000 to 1 million individuals. It is estimated that one-third of people with aniridia actually have WAGR syndrome. Approximately 7 in 1,000 cases of Wilms tumor can be attributed to WAGR syndrome.
Causes
WAGR syndrome is caused by a deletion of genetic material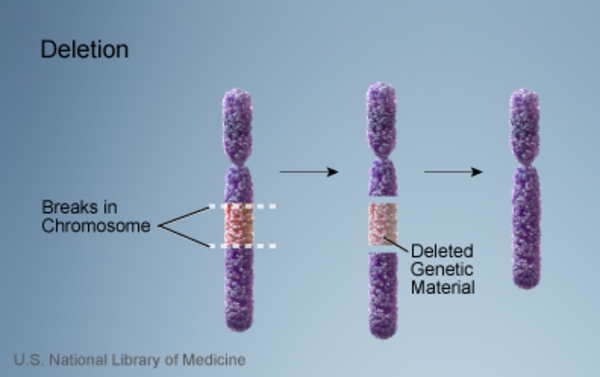 on the short (p) arm
on the short (p) arm of chromosome 11. The size of the deletion varies among affected individuals.
of chromosome 11. The size of the deletion varies among affected individuals.
The signs and symptoms of WAGR syndrome are related to the loss of multiple genes on the short arm of chromosome 11. WAGR syndrome is often described as a contiguous gene deletion syndrome because it is caused by the loss of several neighboring genes.
The PAX6 and WT1 genes are always deleted in people with the typical signs and symptoms of this disorder. Because changes in the PAX6 gene can affect eye development, researchers think that the loss of the PAX6 gene causes the characteristic eye features of WAGR syndrome. The PAX6 gene may also affect brain development.
Variants (also called mutations) in the WT1 gene can cause Wilms tumor and genitourinary abnormalities, so deletion of the WT1 gene is very likely the cause of these features in people with WAGR syndrome.
abnormalities, so deletion of the WT1 gene is very likely the cause of these features in people with WAGR syndrome.
In people with WAGRO syndrome, an additional gene is deleted from chromosome 11. The BDNF gene is active (expressed) in the brain and plays a role in the survival of nerve cells (neurons ). The protein produced from the BDNF gene is thought to be involved in the management of eating, drinking, and body weight. Loss of the BDNF gene is likely the cause of childhood-onset obesity in people with WAGRO syndrome.
). The protein produced from the BDNF gene is thought to be involved in the management of eating, drinking, and body weight. Loss of the BDNF gene is likely the cause of childhood-onset obesity in people with WAGRO syndrome.
People with WAGRO syndrome may be at greater risk of neurological problems such as intellectual disability and autism than those with WAGR syndrome. It is unclear whether this increased risk is due to the loss of the BDNF gene or other nearby genes.
Researchers are still studying the genes that are deleted in people with WAGR syndrome and determining how the loss of these genes causes features of the disorder.
Inheritance
Most cases of WAGR syndrome are not inherited. They are caused by a chromosomal deletion that occurs randomly during the formation of reproductive cells (eggs or sperm) or in early fetal development. Affected people typically have no history of the disorder in their family.
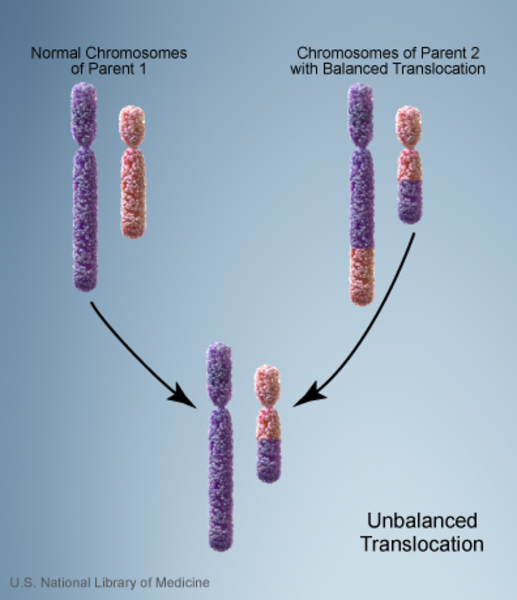
In rare cases, WAGR syndrome is inherited. In these cases, a parent who does not have the signs and symptoms of the condition has a chromosomal rearrangement called a balanced translocation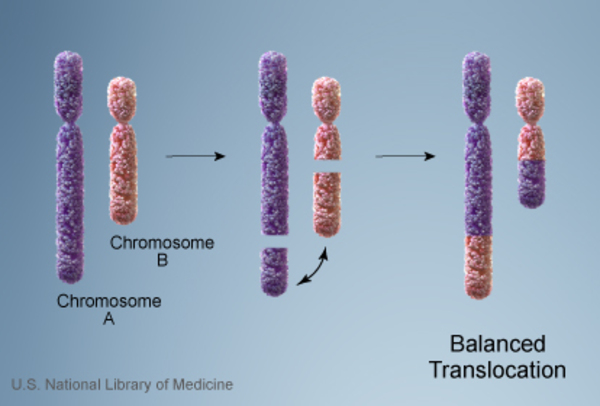 . In this rearrangement, a piece of chromosome 11 is relocated to another chromosome, but no genetic material is gained or lost.
. In this rearrangement, a piece of chromosome 11 is relocated to another chromosome, but no genetic material is gained or lost.
However, the translocation can become unbalanced when genetic material is passed to the next generation. A child may inherit a copy of chromosome 11 this is missing a piece of the short arm, which causes WAGR syndrome.
Other Names for This Condition
- 11p deletion syndrome
- 11p partial monosomy syndrome
- WAGR complex
- WAGR contiguous gene syndrome
- WAGR spectrum disorder
- Wilms tumor, aniridia, genitourinary anomalies, and mental retardation syndrome
- Wilms tumor-aniridia-genital anomalies-retardation syndrome
- Wilms tumor-aniridia-genitourinary anomalies-MR syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Breslow NE, Norris R, Norkool PA, Kang T, Beckwith JB, Perlman EJ, Ritchey ML, Green DM, Nichols KE; National Wilms Tumor Study Group. Characteristics and outcomes of children with the Wilms tumor-Aniridia syndrome: a report from the National Wilms Tumor Study Group. J Clin Oncol. 2003 Dec 15;21(24):4579-85. doi: 10.1200/JCO.2003.06.096. Citation on PubMed
- Duffy KA, Trout KL, Gunckle JM, Krantz SM, Morris J, Kalish JM. Results From the WAGR Syndrome Patient Registry: Characterization of WAGR Spectrum and Recommendations for Care Management. Front Pediatr. 2021 Dec 14;9:733018. doi: 10.3389/fped.2021.733018. eCollection 2021. Citation on PubMed
- Fischbach BV, Trout KL, Lewis J, Luis CA, Sika M. WAGR syndrome: a clinical review of 54 cases. Pediatrics. 2005 Oct;116(4):984-8. doi: 10.1542/peds.2004-0467. Citation on PubMed
- Han JC, Liu QR, Jones M, Levinn RL, Menzie CM, Jefferson-George KS, Adler-Wailes DC, Sanford EL, Lacbawan FL, Uhl GR, Rennert OM, Yanovski JA. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N Engl J Med. 2008 Aug 28;359(9):918-27. doi: 10.1056/NEJMoa0801119. Citation on PubMed or Free article on PubMed Central
- Han JC, Thurm A, Golden Williams C, Joseph LA, Zein WM, Brooks BP, Butman JA, Brady SM, Fuhr SR, Hicks MD, Huey AE, Hanish AE, Danley KM, Raygada MJ, Rennert OM, Martinowich K, Sharp SJ, Tsao JW, Swedo SE. Association of brain-derived neurotrophic factor (BDNF) haploinsufficiency with lower adaptive behaviour and reduced cognitive functioning in WAGR/11p13 deletion syndrome. Cortex. 2013 Nov-Dec;49(10):2700-10. doi: 10.1016/j.cortex.2013.02.009. Epub 2013 Feb 19. Citation on PubMed or Free article on PubMed Central
- Robinson DO, Howarth RJ, Williamson KA, van Heyningen V, Beal SJ, Crolla JA. Genetic analysis of chromosome 11p13 and the PAX6 gene in a series of 125 cases referred with aniridia. Am J Med Genet A. 2008 Mar 1;146A(5):558-69. doi: 10.1002/ajmg.a.32209. Citation on PubMed
- Rodriguez-Lopez R, Perez JM, Balsera AM, Rodriguez GG, Moreno TH, Garcia de Caceres M, Serrano MG, Freijo FC, Ruiz JR, Angueira FB, Perez PM, Estevez MN, Gomez EG. The modifier effect of the BDNF gene in the phenotype of the WAGRO syndrome. Gene. 2013 Mar 10;516(2):285-90. doi: 10.1016/j.gene.2012.11.073. Epub 2012 Dec 21. Citation on PubMed
- Xu S, Han JC, Morales A, Menzie CM, Williams K, Fan YS. Characterization of 11p14-p12 deletion in WAGR syndrome by array CGH for identifying genes contributing to mental retardation and autism. Cytogenet Genome Res. 2008;122(2):181-7. doi: 10.1159/000172086. Epub 2008 Dec 18. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.