

Description
Nonsyndromic hearing loss is a partial or total loss of hearing that is not associated with other signs and symptoms. In contrast, syndromic hearing loss occurs along with signs and symptoms that affect other parts of the body.
Nonsyndromic hearing loss can be classified in several different ways. One common way is by the condition's pattern of inheritance: autosomal dominant (DFNA), autosomal recessive (DFNB), X-linked (DFNX), or mitochondrial. Each of these types of hearing loss includes multiple subtypes. DFNA, DFNB, and DFNX subtypes are numbered in the order in which they were first described. For example, DFNA1 was the first subtype of autosomal dominant nonsyndromic hearing loss to be identified. Sometimes, the subtypes are further broken down using additional letters, as in DFNB1A and DFNB1B.
The characteristics of nonsyndromic hearing loss vary among the different types. Typically, nonsyndromic hearing loss affects both ears (bilateral). The degrees of hearing loss range from mild (difficulty understanding soft speech) to profound (inability to hear even very loud noises). The term "deafness" is often used to describe a nearly total to total (severe-to-profound) hearing loss.
Hearing loss can be stable, or it may be progressive, becoming more severe as a person gets older. Particular types of nonsyndromic hearing loss show distinctive patterns of hearing loss. For example, they can affect the ability to hear high, middle, or low tones.
Most forms of nonsyndromic hearing loss are described as sensorineural, which means they are associated with a permanent loss of hearing that is caused by damage to structures in the inner ear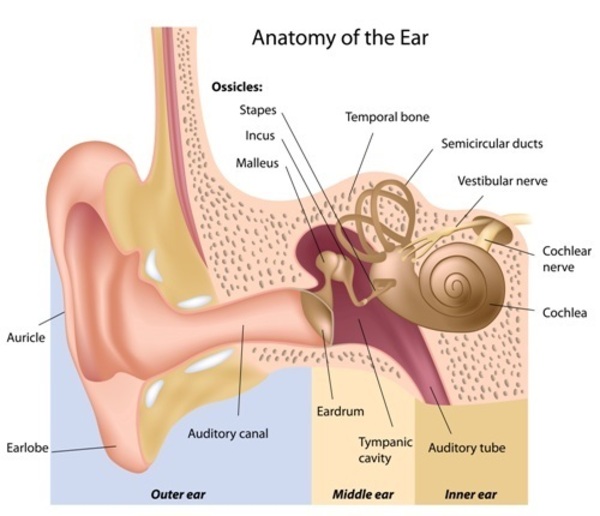 . The inner ear processes sound and sends the information to the brain in the form of electrical nerve impulses. Much less commonly, nonsyndromic hearing loss is described as conductive, meaning it results from changes in the middle ear. The middle ear contains three tiny bones that help transfer sound from the eardrum to the inner ear.
. The inner ear processes sound and sends the information to the brain in the form of electrical nerve impulses. Much less commonly, nonsyndromic hearing loss is described as conductive, meaning it results from changes in the middle ear. The middle ear contains three tiny bones that help transfer sound from the eardrum to the inner ear.
Some forms of nonsyndromic hearing loss involve changes in both the inner ear and the middle ear. This combination is called mixed hearing loss.
Nonsyndromic hearing loss can become apparent at any time throughout life. Hearing loss that is present at birth is described as congenital. Hearing loss that occurs before a child learns to speak is classified as prelingual; if it occurs after the development of speech, it is classified as postlingual.
Frequency
Between 2 and 3 per 1,000 children in the United States are born with measurable hearing loss in 1 or both ears. By age 12, the prevalence of hearing loss increases to 1 in 8 people in the United States.
Genetic factors cause about half of all cases of hearing loss, and 70 percent of genetic-related hearing loss is nonsyndromic.
Causes
The causes of nonsyndromic hearing loss are complex. Researchers have identified more than 155 genes that, when altered, are associated with nonsyndromic hearing loss. However, not all of these associations are equally well understood.
Many of the genes that are altered in people with nonsyndromic hearing loss are involved in the development and function of the inner ear. Genetic changes that contribute to disease are called pathogenic variants. Pathogenic variants in these genes contribute to hearing loss by interfering with critical steps in processing sound. Different pathogenic variants in the same gene can be associated with different types of hearing loss. While a few altered genes are associated with most cases of nonsyndromic hearing loss, variants in many other genes have only been found in one or a few families. In many affected families, the factors that contribute to hearing loss have not been identified.
In about 80 percent of cases, nonsyndromic hearing loss is classified as autosomal recessive. Pathogenic variants in more than 85 genes have been found to cause autosomal recessive nonsyndromic hearing loss. About half of all autosomal recessive nonsyndromic hearing loss results from variants in the GJB2 gene, although this varies by population. When nonsyndromic hearing loss is caused by GJB2 gene variants, it is called DFNB1A. Variants in the STRC gene are the second most common cause of autosomal recessive nonsyndromic hearing loss. These variants cause a form of the condition known as DFNB16.
In about 19 percent of cases, nonsyndromic hearing loss is classified as autosomal dominant. Pathogenic variants in more than 60 genes have been found to cause autosomal dominant nonsyndromic hearing loss. Variants in some of these genes (including GJB2) can also cause autosomal recessive forms of the condition. Pathogenic variants in the KCNQ4 and TECTA genes are among the most common causes of autosomal dominant nonsyndromic hearing loss.
X-linked and mitochondrial forms of nonsyndromic hearing loss are rare, occurring in about 1 percent of cases. About half of all X-linked cases are caused by pathogenic variants in the POU3F4 gene. This form of the condition is designated DFNX2 and often causes conductive hearing loss. Pathogenic variants in at least three other genes have also been identified in people with X-linked nonsyndromic hearing loss.
Mitochondrial forms of hearing loss result from changes in mitochondrial DNA (mtDNA). Mitochondria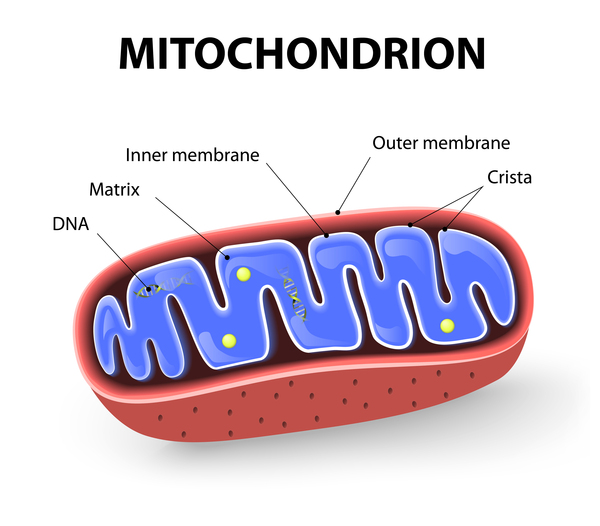 are structures within cells that convert the energy from food into a form that cells can use. Although most DNA is packaged in chromosomes within the nucleus, mitochondria also have a small amount of their own DNA. In rare cases, pathogenic variants in a few mtDNA genes have been associated with nonsyndromic hearing loss.
are structures within cells that convert the energy from food into a form that cells can use. Although most DNA is packaged in chromosomes within the nucleus, mitochondria also have a small amount of their own DNA. In rare cases, pathogenic variants in a few mtDNA genes have been associated with nonsyndromic hearing loss.
Pathogenic variants in some of the genes that are associated with nonsyndromic hearing loss can also cause syndromic forms of hearing loss. It is often unclear how variants in the same gene can cause isolated hearing loss in some individuals and hearing loss with additional signs and symptoms in others. It is estimated that about 20 percent of children with nonsyndromic hearing loss will develop additional features and later receive a diagnosis of syndromic hearing loss.
In addition to genetic changes, hearing loss can be caused by environmental factors or a combination of genetic risk and a person's environmental exposures. Environmental causes of hearing loss include certain medications, specific infections before or after birth, and exposure to loud noise over an extended period. Age is also a major risk factor for hearing loss. Age-related hearing loss (presbycusis) has both genetic and environmental influences.
Inheritance
Nonsyndromic hearing loss can be inherited in different ways.
Autosomal recessive nonsyndromic hearing loss occurs when both copies of the gene in each cell have a pathogenic variant. Typically, each parent of an individual with autosomal recessive nonsyndromic hearing loss carries one copy of the altered gene but does not have hearing loss.
nonsyndromic hearing loss occurs when both copies of the gene in each cell have a pathogenic variant. Typically, each parent of an individual with autosomal recessive nonsyndromic hearing loss carries one copy of the altered gene but does not have hearing loss.
Autosomal dominant nonsyndromic hearing loss occurs when one copy of the altered gene in each cell is sufficient to cause the condition. Most people with autosomal dominant nonsyndromic hearing loss inherit an altered copy of the gene from a parent who also has hearing loss.
nonsyndromic hearing loss occurs when one copy of the altered gene in each cell is sufficient to cause the condition. Most people with autosomal dominant nonsyndromic hearing loss inherit an altered copy of the gene from a parent who also has hearing loss.
X-linked nonsyndromic hearing loss occurs when the altered gene that causes the disorder is located on the X chromosome, which is one of the two sex chromosomes in each cell. Men and boys with X-linked nonsyndromic hearing loss tend to develop more severe hearing loss earlier in life than women and girls who inherit a copy of the same variant. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
in each cell. Men and boys with X-linked nonsyndromic hearing loss tend to develop more severe hearing loss earlier in life than women and girls who inherit a copy of the same variant. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Mitochondrial nonsyndromic hearing loss is caused by changes to mtDNA.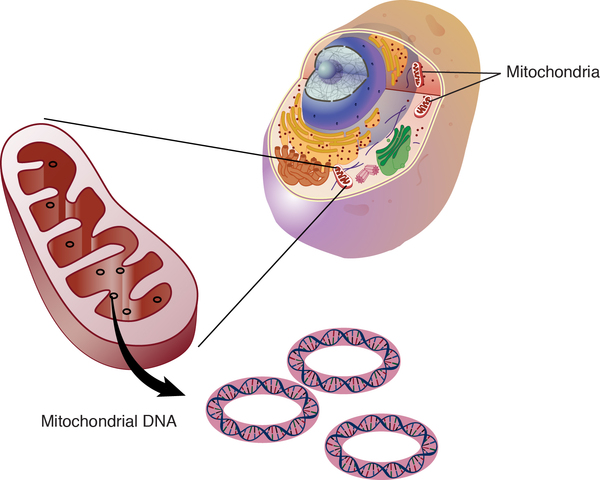 These cases are inherited in a mitochondrial pattern
These cases are inherited in a mitochondrial pattern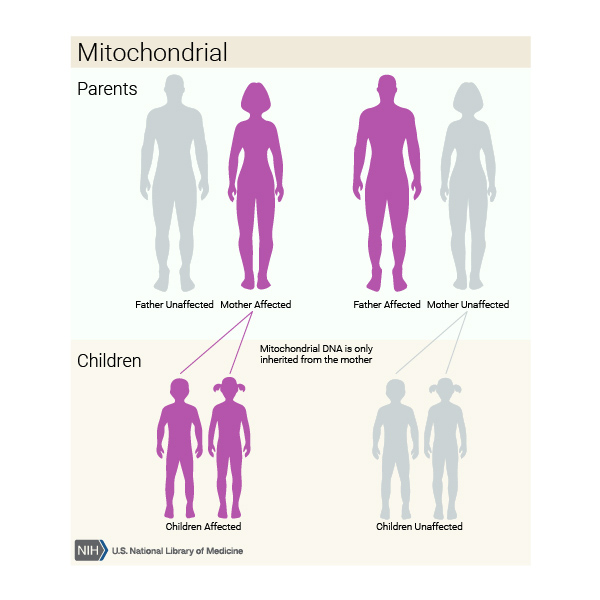 , which is also known as maternal inheritance. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can only inherit disorders that are caused by mtDNA variants from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits that are associated with changes in mtDNA to their children.
, which is also known as maternal inheritance. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can only inherit disorders that are caused by mtDNA variants from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits that are associated with changes in mtDNA to their children.
In some cases, hearing loss occurs in people with no history of the condition in their family. These cases are described as sporadic, and the cause of hearing loss is often unknown. When hearing loss is caused by environmental factors, it is not inherited.
Other Names for This Condition
- Isolated deafness
- Nonsyndromic deafness
- Nonsyndromic hearing impairment
- Nonsyndromic hearing loss and deafness
Additional Information & Resources
Patient Support and Advocacy Resources
Clinical Trials
Scientific Articles on PubMed
References
- Del Castillo I, Morin M, Dominguez-Ruiz M, Moreno-Pelayo MA. Genetic etiology of non-syndromic hearing loss in Europe. Hum Genet. 2022 Apr;141(3-4):683-696. doi: 10.1007/s00439-021-02425-6. Epub 2022 Jan 19. Citation on PubMed
- Ding Y, Leng J, Fan F, Xia B, Xu P. The role of mitochondrial DNA mutations in hearing loss. Biochem Genet. 2013 Aug;51(7-8):588-602. doi: 10.1007/s10528-013-9589-6. Epub 2013 Apr 21. Citation on PubMed
- Lezirovitz K, Mingroni-Netto RC. Genetic etiology of non-syndromic hearing loss in Latin America. Hum Genet. 2022 Apr;141(3-4):539-581. doi: 10.1007/s00439-021-02354-4. Epub 2021 Oct 15. Citation on PubMed
- Lieu JEC, Kenna M, Anne S, Davidson L. Hearing Loss in Children: A Review. JAMA. 2020 Dec 1;324(21):2195-2205. doi: 10.1001/jama.2020.17647. Citation on PubMed
- Morris JA, Gonzalez T, Blanton SH, Angeli SI, Liu XZ. GJB2-Related Hearing Loss: Genotype-Phenotype Correlations, Natural History, and Emerging Therapeutic Strategies. Int J Mol Sci. 2026 Jan 3;27(1):491. doi: 10.3390/ijms27010491. Citation on PubMed
- Ray M, Sarkar S, Sable MN. Comprehensive functional network analysis and screening of deleterious pathogenic variants in non-syndromic hearing loss causative genes. Biosci Rep. 2021 Oct 29;41(10):BSR20211865. doi: 10.1042/BSR20211865. Citation on PubMed
- Reda Del Barrio S, de Vergas Gutierrez J, Quesada-Espinosa JF, Sanchez-Calvin MT, Gomez-Manjon I, Sierra-Tomillo O, Juarez-Rufian A, Garcia Fernandez A. Diagnostic yield of genetic testing in adults with sensorineural hearing loss. Acta Otorrinolaringol Esp (Engl Ed). 2024 May-Jun;75(3):185-191. doi: 10.1016/j.otoeng.2023.10.007. Epub 2024 Feb 10. Citation on PubMed
- Redfield S, Shearer AE. STRC-Related Autosomal Recessive Hearing Loss. 2023 Dec 14. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK598310/ Citation on PubMed
- Rentas S, Rajagopalan R, Ayazseven T, Sarmady M, Raible SE, Krantz ID, Abou Tayoun AN. Whole Genome Sequencing Improves the Identification of Pathogenic and Novel Variation in Nonsyndromic Hearing Loss. Hum Mutat. 2025 Dec 1;2025:6371082. doi: 10.1155/humu/6371082. eCollection 2025. Citation on PubMed
- Sharma N, Kumari D, Panigrahi I, Khetarpal P. A systematic review of the monogenic causes of Non-Syndromic Hearing Loss (NSHL) and discussion of Current Diagnosis and Treatment options. Clin Genet. 2023 Jan;103(1):16-34. doi: 10.1111/cge.14228. Epub 2022 Sep 29. Citation on PubMed
- Shearer AE, Hildebrand MS, Odell AM, Smith RJH. Genetic Hearing Loss Overview. 1999 Feb 14 [updated 2025 Apr 3]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1434/ Citation on PubMed
- Smith RJH, Azaiez H, Booth K. GJB2-Related Autosomal Recessive Nonsyndromic Hearing Loss. 1998 Sep 28 [updated 2023 Jul 20]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1272/ Citation on PubMed
- Smith RJH, Hildebrand M. DFNA2 Nonsyndromic Hearing Loss. 2008 Apr 4 [updated 2018 May 10]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1209/ Citation on PubMed
- Usami SI, Nishio SY. Nonsyndromic Hearing Loss and Deafness, Mitochondrial. 2004 Oct 22 [updated 2018 Jun 14]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1422/ Citation on PubMed
- Young A, Ng M. Genetic Hearing Loss. 2023 Apr 17. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from http://www.ncbi.nlm.nih.gov/books/NBK580517/ Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.