

Description
Xeroderma pigmentosum, commonly known as XP, is an inherited condition characterized by an extreme sensitivity to ultraviolet radiation (UVR) , which is present in sunlight and may also be found in some types of artificial lighting. This condition mostly affects the eyes and areas of skin exposed to the sun. Xeroderma pigmentosum is associated with an increased risk of UVR-induced cancers. People with this condition often experience premature aging. Some affected individuals also have problems involving the nervous system.
, which is present in sunlight and may also be found in some types of artificial lighting. This condition mostly affects the eyes and areas of skin exposed to the sun. Xeroderma pigmentosum is associated with an increased risk of UVR-induced cancers. People with this condition often experience premature aging. Some affected individuals also have problems involving the nervous system.
The signs of xeroderma pigmentosum usually appear in infancy or early childhood. About half of affected children develop a severe sunburn after spending just a few minutes in the sun. The sunburn causes redness and blistering that can last for weeks. However, some children with xeroderma pigmentosum can tan normally.
By age 2, almost all children with xeroderma pigmentosum develop freckling of the skin in sun-exposed areas (such as the face, arms, and lips); this type of freckling rarely occurs in young children without the disorder. In affected individuals, exposure to sunlight often causes dry skin (xeroderma) and changes in skin coloring (pigmentation). This combination of features gives the condition its name.
People with xeroderma pigmentosum are 10,000 times more likely to develop non-melanoma skin cancer and up to 2,000 times more likely to develop melanoma skin cancer compared to individuals without this condition. The types of skin cancer that can develop include basal cell carcinoma
and up to 2,000 times more likely to develop melanoma skin cancer compared to individuals without this condition. The types of skin cancer that can develop include basal cell carcinoma , squamous cell carcinoma
, squamous cell carcinoma , and melanoma. Most commonly, the first skin cancer appears in affected individuals before age 10.
, and melanoma. Most commonly, the first skin cancer appears in affected individuals before age 10.
Without protection from the sun and other sources of UVR, most people with xeroderma pigmentosum develop multiple skin cancers during their lifetime. These cancers occur most often on portions of the body that are exposed to the sun, including the face, the lips, the eyelids, the surface of the eyes, the scalp, and the tip of the tongue. Studies suggest that people with xeroderma pigmentosum may also have an increased risk of some internal cancers, including brain tumors, thyroid cancer , and blood cancers. Additionally, affected individuals who smoke cigarettes have a significantly increased risk of lung cancer.
, and blood cancers. Additionally, affected individuals who smoke cigarettes have a significantly increased risk of lung cancer.
The eyes of people with xeroderma pigmentosum may be painfully sensitive to UVR (photophobia). If the eyes are not protected from UVR, they may become bloodshot and irritated, and the clear front covering of the eyes (the cornea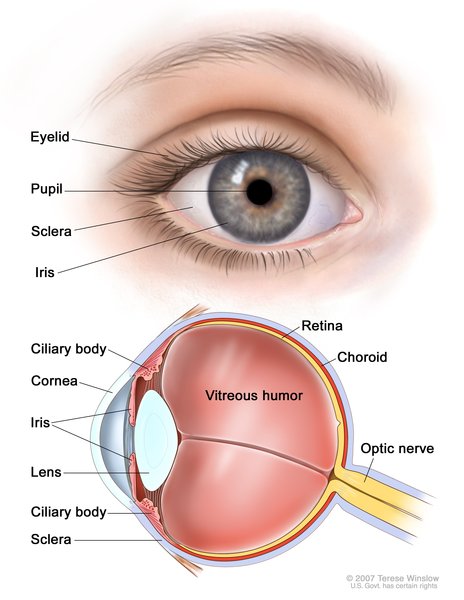 ) may become cloudy. In some people, the eyelashes fall out and the eyelids may be thin and turn abnormally inward or outward. In addition to an increased risk of cancer on the surface of the eye, xeroderma pigmentosum is associated with noncancerous growths on the eye. Many of these eye abnormalities can impair vision.
) may become cloudy. In some people, the eyelashes fall out and the eyelids may be thin and turn abnormally inward or outward. In addition to an increased risk of cancer on the surface of the eye, xeroderma pigmentosum is associated with noncancerous growths on the eye. Many of these eye abnormalities can impair vision.
About 30 percent of people with xeroderma pigmentosum develop progressive neurological abnormalities in addition to problems involving the skin and eyes. These abnormalities can include hearing loss, poor coordination, difficulty walking, movement problems, loss of intellectual function, difficulty swallowing and talking, and seizures. When these neurological problems occur, they tend to worsen with time.
Individuals with xeroderma pigmentosum may experience early menopause.
Researchers have identified at least eight genetic forms of xeroderma pigmentosum : complementation group A (XP-A) through complementation group G (XP-G), plus a variant type (XP-V). The types are distinguished by their genetic cause. All of the types increase the risk of skin cancer, although some are more likely than others to be associated with neurological abnormalities.
: complementation group A (XP-A) through complementation group G (XP-G), plus a variant type (XP-V). The types are distinguished by their genetic cause. All of the types increase the risk of skin cancer, although some are more likely than others to be associated with neurological abnormalities.
Frequency
Xeroderma pigmentosum is a rare disorder; it is estimated to affect about 1 in 1 million people in the United States and Europe. The condition is more common in Japan, North Africa, and the Middle East.
Causes
Xeroderma pigmentosum is caused by variants (also called mutations) in at least nine genes: DDB2, ERCC1, ERCC2, ERCC3, ERCC4, ERCC5, POLH, XPA, and XPC. These genes are involved in repairing damaged DNA. DNA can be damaged by UVR and by toxic chemicals, such as those found in cigarette smoke. Normal cells are usually able to fix DNA damage before it causes problems. However, in people with xeroderma pigmentosum, DNA damage is not repaired normally. As damage builds up in DNA, cells malfunction and eventually become cancerous or die.
can be damaged by UVR and by toxic chemicals, such as those found in cigarette smoke. Normal cells are usually able to fix DNA damage before it causes problems. However, in people with xeroderma pigmentosum, DNA damage is not repaired normally. As damage builds up in DNA, cells malfunction and eventually become cancerous or die.
Many of the genes related to xeroderma pigmentosum are part of a DNA-repair process known as nucleotide excision repair (NER) . The proteins produced from these genes play a variety of roles in this process. They recognize DNA damage, unwind regions of DNA where the damage has occurred, snip out (excise) the abnormal sections, and replace the damaged areas with the correct DNA. Changes in NER-related genes prevent cells from carrying out one or more of these steps. The DDB2, ERCC1, ERCC2, ERCC3, ERCC4, ERCC5, XPA, and XPC genes are all involved in the NER process. The POLH gene also plays a role in protecting cells from UVR-induced DNA damage by bypassing the DNA damage, although it is not involved in NER.
. The proteins produced from these genes play a variety of roles in this process. They recognize DNA damage, unwind regions of DNA where the damage has occurred, snip out (excise) the abnormal sections, and replace the damaged areas with the correct DNA. Changes in NER-related genes prevent cells from carrying out one or more of these steps. The DDB2, ERCC1, ERCC2, ERCC3, ERCC4, ERCC5, XPA, and XPC genes are all involved in the NER process. The POLH gene also plays a role in protecting cells from UVR-induced DNA damage by bypassing the DNA damage, although it is not involved in NER.
More than half of all cases of xeroderma pigmentosum in the United States are caused by variants in the XPC, ERCC2, and POLH genes. Variants in the other genes generally account for a smaller percentage of cases.
The major features of xeroderma pigmentosum result from a buildup of unrepaired DNA damage. When UVR damages genes that control cell growth and division, cells can either die or grow too fast and in an uncontrolled way. Unregulated cell growth can lead to the development of cancerous tumors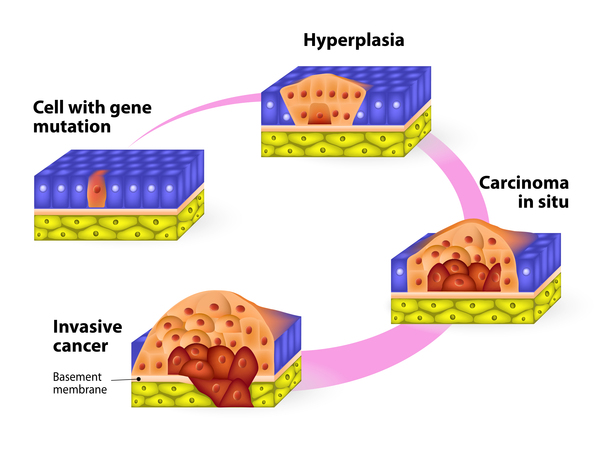 . The neurological abnormalities are also thought to result from a builup of DNA damage, although the brain is not exposed to UVR. Researchers suspect that other factors damage DNA in nerve cells. It is unclear why some people with xeroderma pigmentosum develop neurological abnormalities and others do not.
. The neurological abnormalities are also thought to result from a builup of DNA damage, although the brain is not exposed to UVR. Researchers suspect that other factors damage DNA in nerve cells. It is unclear why some people with xeroderma pigmentosum develop neurological abnormalities and others do not.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- DeSanctis-Cacchione syndrome
- XP
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: ERCC1-Related Xeroderma Pigmentosum

- Genetic Testing Registry: Xeroderma pigmentosum group B
- Genetic Testing Registry: Xeroderma pigmentosum
- Genetic Testing Registry: Xeroderma pigmentosum group A
- Genetic Testing Registry: Xeroderma pigmentosum variant type
- Genetic Testing Registry: Xeroderma pigmentosum, group C
- Genetic Testing Registry: Xeroderma pigmentosum, group D
- Genetic Testing Registry: Xeroderma pigmentosum, group E
- Genetic Testing Registry: Xeroderma pigmentosum, group F
- Genetic Testing Registry: Xeroderma pigmentosum, group G
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- XERODERMA PIGMENTOSUM, COMPLEMENTATION GROUP A; XPA
- XERODERMA PIGMENTOSUM, COMPLEMENTATION GROUP C; XPC
- XERODERMA PIGMENTOSUM, COMPLEMENTATION GROUP D; XPD
- XERODERMA PIGMENTOSUM, COMPLEMENTATION GROUP E; XPE
- XERODERMA PIGMENTOSUM, VARIANT TYPE; XPV
- XERODERMA PIGMENTOSUM, COMPLEMENTATION GROUP F; XPF
- XERODERMA PIGMENTOSUM, COMPLEMENTATION GROUP G; XPG
- XERODERMA PIGMENTOSUM, COMPLEMENTATION GROUP B; XPB
Scientific Articles on PubMed
References
- DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol. 2012 Mar;132(3 Pt 2):785-96. doi: 10.1038/jid.2011.426. Epub 2012 Jan 5. Citation on PubMed
- Kleijer WJ, Laugel V, Berneburg M, Nardo T, Fawcett H, Gratchev A, Jaspers NG, Sarasin A, Stefanini M, Lehmann AR. Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst). 2008 May 3;7(5):744-50. doi: 10.1016/j.dnarep.2008.01.014. Epub 2008 Mar 10. Citation on PubMed
- Kraemer KH, DiGiovanna JJ, Tamura D. Xeroderma Pigmentosum. 2003 Jun 20 [updated 2022 Mar 24]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1397/ Citation on PubMed
- Merideth M, Tamura D, Angra D, Khan SG, Ferrell J, Goldstein AM, DiGiovanna JJ, Kraemer KH. Reproductive Health in Xeroderma Pigmentosum: Features of Premature Aging. Obstet Gynecol. 2019 Oct;134(4):814-819. doi: 10.1097/AOG.0000000000003490. Citation on PubMed
- Nikolaev S, Yurchenko AA, Sarasin A. Increased risk of internal tumors in DNA repair-deficient xeroderma pigmentosum patients: analysis of four international cohorts. Orphanet J Rare Dis. 2022 Mar 4;17(1):104. doi: 10.1186/s13023-022-02203-1. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.