

Description
Wolfram syndrome is a condition that affects many of the body's systems. The hallmark features of Wolfram syndrome are high blood sugar (glucose) levels resulting from a shortage of the hormone insulin (a condition called diabetes mellitus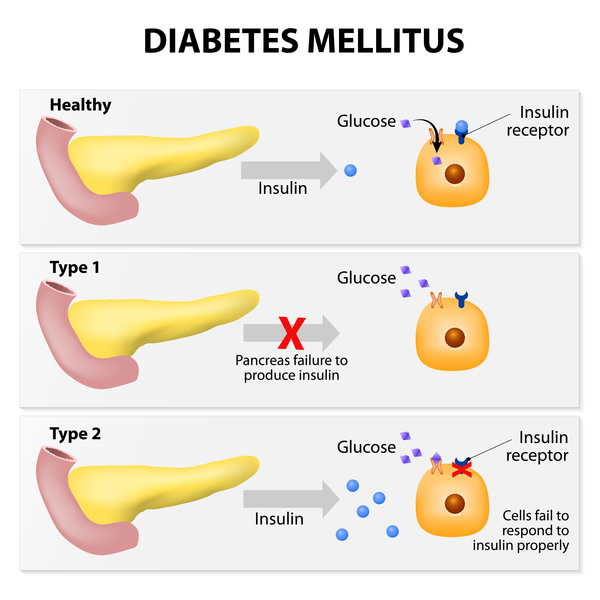 ) and progressive vision loss due to degeneration of the nerves that carry information from the eyes
) and progressive vision loss due to degeneration of the nerves that carry information from the eyes to the brain (a condition called optic atrophy). People with Wolfram syndrome often also have pituitary gland dysfunction that results in excess urine production (a condition called diabetes insipidus), hearing loss caused by changes in the inner ear
to the brain (a condition called optic atrophy). People with Wolfram syndrome often also have pituitary gland dysfunction that results in excess urine production (a condition called diabetes insipidus), hearing loss caused by changes in the inner ear (sensorineural deafness), urinary tract problems, reduced amounts of the sex hormone testosterone in males (hypogonadism), or neurological or psychiatric disorders.
(sensorineural deafness), urinary tract problems, reduced amounts of the sex hormone testosterone in males (hypogonadism), or neurological or psychiatric disorders.
Diabetes mellitus is typically the first symptom of Wolfram syndrome, usually diagnosed around age 6. Nearly everyone with Wolfram syndrome who develops diabetes mellitus requires insulin replacement therapy. Optic atrophy is often the next symptom to appear, usually around age 11. The first signs of optic atrophy are loss of color vision and side (peripheral) vision. Over time, the vision problems get worse, and people with optic atrophy are usually blind within approximately 8 years after signs of optic atrophy first begin.
In diabetes insipidus, the pituitary gland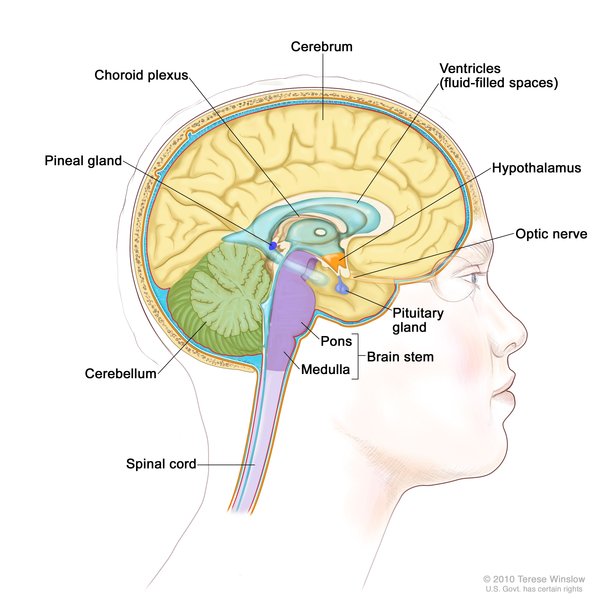 , which is located at the base of the brain, does not function normally. This abnormality disrupts the release of a hormone called vasopressin, which helps control the body's water balance and urine production. Approximately 70 percent of people with Wolfram syndrome have diabetes insipidus. Pituitary gland dysfunction can also cause hypogonadism in males. The lack of testosterone that occurs with hypogonadism affects growth and sexual development. About 65 percent of people with Wolfram syndrome have sensorineural deafness that can range in severity from deafness beginning at birth to mild hearing loss beginning in adolescence that worsens over time. Sixty to 90 percent of people with Wolfram syndrome have a urinary tract problem. Urinary tract problems include obstruction of the ducts between the kidneys and bladder
, which is located at the base of the brain, does not function normally. This abnormality disrupts the release of a hormone called vasopressin, which helps control the body's water balance and urine production. Approximately 70 percent of people with Wolfram syndrome have diabetes insipidus. Pituitary gland dysfunction can also cause hypogonadism in males. The lack of testosterone that occurs with hypogonadism affects growth and sexual development. About 65 percent of people with Wolfram syndrome have sensorineural deafness that can range in severity from deafness beginning at birth to mild hearing loss beginning in adolescence that worsens over time. Sixty to 90 percent of people with Wolfram syndrome have a urinary tract problem. Urinary tract problems include obstruction of the ducts between the kidneys and bladder (ureters), a large bladder that cannot empty normally (high-capacity atonal bladder), disrupted urination (bladder sphincter dyssynergia), and difficulty controlling the flow of urine (incontinence).
(ureters), a large bladder that cannot empty normally (high-capacity atonal bladder), disrupted urination (bladder sphincter dyssynergia), and difficulty controlling the flow of urine (incontinence).
About 60 percent of people with Wolfram syndrome develop a neurological or psychiatric disorder, most commonly problems with balance and coordination (ataxia), typically beginning in early adulthood. Other neurological problems experienced by people with Wolfram syndrome include irregular breathing caused by the brain's inability to control breathing (central apnea), loss of the sense of smell (anosmia), loss of the gag reflex, muscle spasms (myoclonus), seizures, reduced sensation in the lower extremities (peripheral neuropathy), and intellectual impairment. Psychiatric disorders associated with Wolfram syndrome include psychosis, episodes of severe depression, and impulsive and aggressive behavior.
There are two types of Wolfram syndrome with many overlapping features. The two types are differentiated by their genetic cause. In addition to the usual features of Wolfram syndrome type 1 (described above), individuals with Wolfram syndrome type 2 have stomach or intestinal ulcers and excessive bleeding after an injury. The tendency to bleed excessively combined with the ulcers typically leads to abnormal bleeding in the gastrointestinal system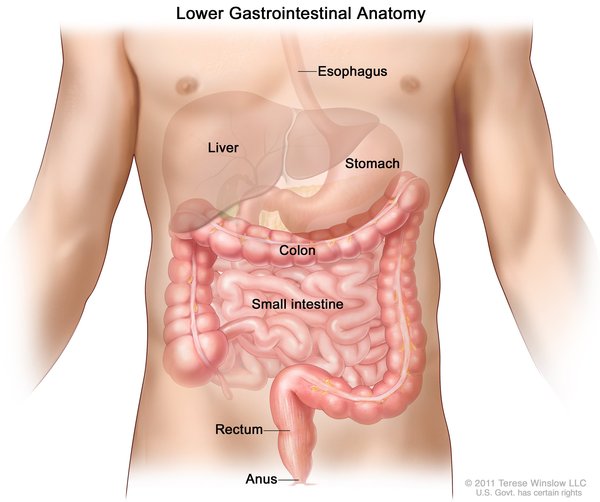 . People with Wolfram syndrome type 2 do not develop diabetes insipidus.
. People with Wolfram syndrome type 2 do not develop diabetes insipidus.
Historically, Wolfram syndrome was fatal by mid-adulthood due to complications from the many features of the condition, such as health problems related to diabetes mellitus or neurological problems. However, with better diagnosis and management, life expectancy has risen.
Frequency
The estimated prevalence of Wolfram syndrome type 1 is 1 in 500,000 people worldwide. Approximately 200 cases have been described in the scientific literature. Only a few families from Jordan have been found to have Wolfram syndrome type 2.
Causes
Variants (also known as mutations) in the WFS1 gene cause more than 90 percent of Wolfram syndrome type 1 cases. This gene provides instructions for producing a protein called wolframin that is thought to regulate the amount of calcium in cells. A proper calcium balance is important for many different cellular functions, including cell-to-cell communication, the tensing (contraction) of muscles, and protein processing. The wolframin protein is found in many different tissues, such as the pancreas, brain, heart, bones, muscles, lung, liver, and kidneys. Within cells, wolframin is located in the membrane of a cell structure called the endoplasmic reticulum that is involved in protein production, processing, and transport. Wolframin's function is important in the pancreas
that is involved in protein production, processing, and transport. Wolframin's function is important in the pancreas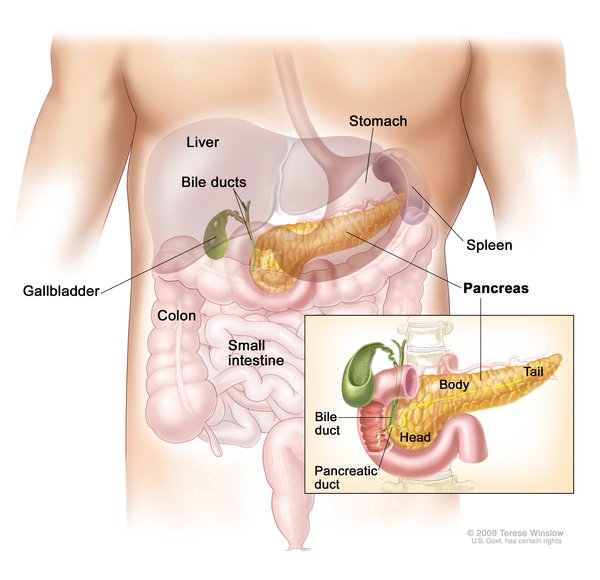 , where the protein is thought to help process a protein called proinsulin
, where the protein is thought to help process a protein called proinsulin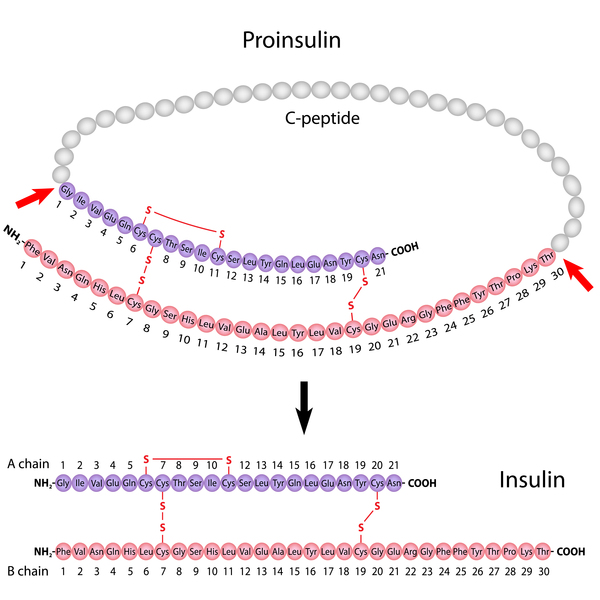 into the mature hormone insulin. This hormone helps control blood glucose levels.
into the mature hormone insulin. This hormone helps control blood glucose levels.
WFS1 gene variants lead to the production of a wolframin protein that has reduced or absent function. As a result, calcium levels within cells are not regulated and the endoplasmic reticulum does not work correctly. When the endoplasmic reticulum does not have enough functional wolframin, the cell triggers its own cell death (apoptosis). The death of cells in the pancreas, specifically cells that make insulin (beta cells), causes diabetes mellitus in people with Wolfram syndrome. The gradual loss of cells along the optic nerve eventually leads to blindness in affected individuals. The death of cells in other body systems likely causes the various signs and symptoms of Wolfram syndrome type 1.
A certain variant in the CISD2 gene was found to cause Wolfram syndrome type 2. The CISD2 gene provides instructions for making a protein that is located in the outer membrane of cell structures called mitochondria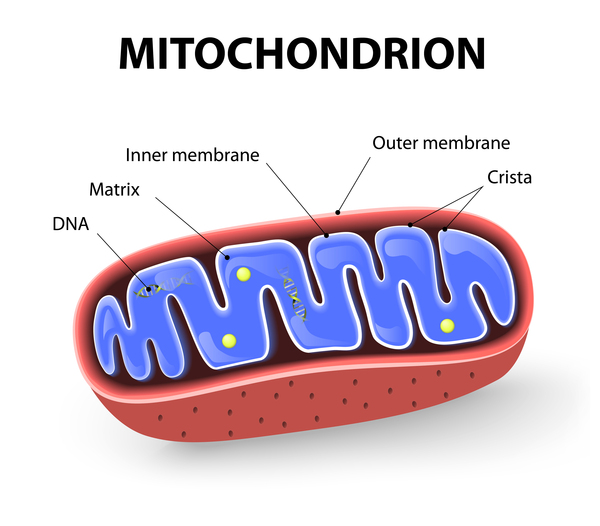 . Mitochondria are the energy-producing centers of cells. The exact function of the CISD2 protein is unknown, but it is thought to help keep mitochondria functioning normally.
. Mitochondria are the energy-producing centers of cells. The exact function of the CISD2 protein is unknown, but it is thought to help keep mitochondria functioning normally.
The CISD2 gene variant that causes Wolfram syndrome type 2 results in an abnormally small, nonfunctional CISD2 protein. As a result, mitochondria are not properly maintained, and they eventually break down. Since the mitochondria provide energy to cells, the loss of mitochondria results in decreased energy for cells. Cells that do not have enough energy to function will eventually die. Cells with high energy demands such as nerve cells in the brain, eye, or gastrointestinal tract are most susceptible to cell death due to reduced energy. It is unknown why people with CISD2 gene variants have ulcers and bleeding problems in addition to the usual Wolfram syndrome features.
Some people with Wolfram syndrome do not have an identified variant in either the WFS1 or CISD2 gene. The cause of the condition in these individuals is unknown.
Inheritance
When Wolfram syndrome is caused by variants in the WFS1 gene, it is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell are altered. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition. Some studies have shown that people who have one copy of a WFS1 gene variant are at increased risk of developing individual features of Wolfram syndrome or related features, such as type 2 diabetes, hearing loss, or psychiatric illness. However, other studies have found no increased risk in such individuals.
, which means both copies of the gene in each cell are altered. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition. Some studies have shown that people who have one copy of a WFS1 gene variant are at increased risk of developing individual features of Wolfram syndrome or related features, such as type 2 diabetes, hearing loss, or psychiatric illness. However, other studies have found no increased risk in such individuals.
Wolfram syndrome caused by variants in the CISD2 gene is also inherited in an autosomal recessive pattern.
Other Names for This Condition
- Diabetes insipidus and mellitus with optic atrophy and deafness
- Diabetes insipidus, diabetes mellitus, optic atrophy, and deafness
- DIDMOAD
- DIDMOAD syndrome
- DIDMOADUD
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Aloi C, Salina A, Pasquali L, Lugani F, Perri K, Russo C, Tallone R, Ghiggeri GM, Lorini R, d'Annunzio G. Wolfram syndrome: new mutations, different phenotype. PLoS One. 2012;7(1):e29150. doi: 10.1371/journal.pone.0029150. Epub 2012 Jan 4. Citation on PubMed or Free article on PubMed Central
- Amr S, Heisey C, Zhang M, Xia XJ, Shows KH, Ajlouni K, Pandya A, Satin LS, El-Shanti H, Shiang R. A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am J Hum Genet. 2007 Oct;81(4):673-83. doi: 10.1086/520961. Epub 2007 Aug 20. Citation on PubMed or Free article on PubMed Central
- Chaussenot A, Bannwarth S, Rouzier C, Vialettes B, Mkadem SA, Chabrol B, Cano A, Labauge P, Paquis-Flucklinger V. Neurologic features and genotype-phenotype correlation in Wolfram syndrome. Ann Neurol. 2011 Mar;69(3):501-8. doi: 10.1002/ana.22160. Epub 2010 Dec 28. Citation on PubMed
- de Heredia ML, Cleries R, Nunes V. Genotypic classification of patients with Wolfram syndrome: insights into the natural history of the disease and correlation with phenotype. Genet Med. 2013 Jul;15(7):497-506. doi: 10.1038/gim.2012.180. Epub 2013 Feb 21. Citation on PubMed
- Kanki T, Klionsky DJ. Mitochondrial abnormalities drive cell death in Wolfram syndrome 2. Cell Res. 2009 Aug;19(8):922-3. doi: 10.1038/cr.2009.94. No abstract available. Citation on PubMed or Free article on PubMed Central
- Marshall BA, Permutt MA, Paciorkowski AR, Hoekel J, Karzon R, Wasson J, Viehover A, White NH, Shimony JS, Manwaring L, Austin P, Hullar TE, Hershey T; Washington University Wolfram Study Group. Phenotypic characteristics of early Wolfram syndrome. Orphanet J Rare Dis. 2013 Apr 27;8:64. doi: 10.1186/1750-1172-8-64. Citation on PubMed or Free article on PubMed Central
- Rigoli L, Di Bella C. Wolfram syndrome 1 and Wolfram syndrome 2. Curr Opin Pediatr. 2012 Aug;24(4):512-7. doi: 10.1097/MOP.0b013e328354ccdf. Citation on PubMed
- Rigoli L, Lombardo F, Di Bella C. Wolfram syndrome and WFS1 gene. Clin Genet. 2011 Feb;79(2):103-17. doi: 10.1111/j.1399-0004.2010.01522.x. Epub 2010 Aug 26. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.