

Description
Williams syndrome is a developmental disorder that affects many parts of the body. This condition is characterized by mild to moderate intellectual disability or learning problems, unique personality characteristics, distinctive facial features, and heart and blood vessel (cardiovascular) problems.
People with Williams syndrome typically have difficulty with visual-spatial tasks such as drawing and assembling puzzles, but they tend to do well on tasks that involve spoken language, music, and learning by repetition (rote memorization). Affected individuals have outgoing, engaging personalities and tend to take an extreme interest in other people. Attention deficit disorder (ADD), problems with anxiety, and phobias are common among people with this disorder.
Young children with Williams syndrome have distinctive facial features including a broad forehead, puffiness around the eyes, a flat bridge of the nose, full cheeks, and a small chin. Many affected people have dental problems such as teeth that are small, widely spaced, crooked, or missing. Older children and adults typically have a longer face with a wide mouth and full lips.
A form of cardiovascular disease called supravalvular aortic stenosis (SVAS) occurs frequently in people with Williams syndrome. Supravalvular aortic stenosis is a narrowing of the large blood vessel that carries blood from the heart to the rest of the body (the aorta ). If this condition is not treated, the aortic narrowing can lead to shortness of breath, chest pain, and heart failure. Narrowing of other vessels, including the artery from the heart to the lungs (pulmonary stenosis) and the arteries that supply blood to the heart (coronary artery stenosis) can also occur. Other problems with the heart and blood vessels, including high blood pressure (hypertension) and stiff blood vessels, have also been reported in people with Williams syndrome. Individuals with Williams syndrome have an increased risk of complications with the use of anesthesia.
). If this condition is not treated, the aortic narrowing can lead to shortness of breath, chest pain, and heart failure. Narrowing of other vessels, including the artery from the heart to the lungs (pulmonary stenosis) and the arteries that supply blood to the heart (coronary artery stenosis) can also occur. Other problems with the heart and blood vessels, including high blood pressure (hypertension) and stiff blood vessels, have also been reported in people with Williams syndrome. Individuals with Williams syndrome have an increased risk of complications with the use of anesthesia.
Additional signs and symptoms of Williams syndrome include abnormalities of connective tissue (tissue that supports the body's joints and organs) such as joint problems and soft, loose skin. Affected people may also have increased calcium levels in the blood (hypercalcemia) in infancy, developmental delays, problems with coordination, and short stature. Medical problems involving vision or hearing, including sensitivity to sound (hyperacusis), are frequently associated with Williams syndrome. In addition, problems with the digestive tract and the urinary system
and the urinary system are also possible. Obesity or diabetes can develop in adulthood.
are also possible. Obesity or diabetes can develop in adulthood.
Frequency
Williams syndrome affects an estimated 1 in 7,500 to 18,000 people.
Causes
Williams syndrome is caused by the loss (deletion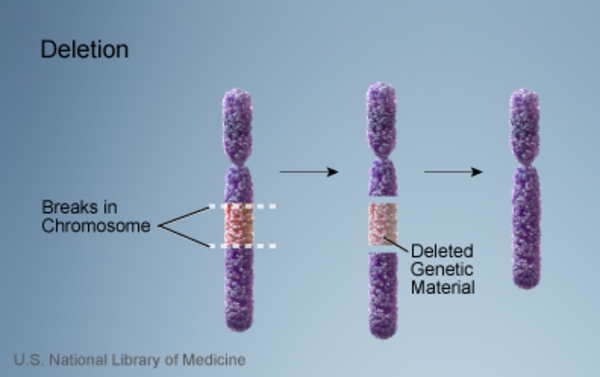 ) of genetic material from a specific region of chromosome 7. The deleted region includes 25 to 27 genes, and researchers believe that a loss of several of these genes contributes to the characteristic features of this disorder.
) of genetic material from a specific region of chromosome 7. The deleted region includes 25 to 27 genes, and researchers believe that a loss of several of these genes contributes to the characteristic features of this disorder.
ELN, GTF2I, GTF2IRD1, and LIMK1 are among the genes that are typically deleted in people with Williams syndrome. Researchers have found that loss of the ELN gene is associated with the connective tissue abnormalities and cardiovascular disease (specifically supravalvular aortic stenosis) found in many people with this disease. Studies suggest that deletion of GTF2I, GTF2IRD1, LIMK1, and perhaps other genes may help explain the characteristic difficulties with visual-spatial tasks, unique behavioral characteristics, and other cognitive difficulties seen in people with Williams syndrome. Loss of the GTF2IRD1 gene may also contribute to the distinctive facial features often associated with this condition.
Researchers believe that the presence or absence of the NCF1 gene on chromosome 7 impacts the risk of developing hypertension in people with Williams syndrome. When the NCF1 gene is included in the part of the chromosome that is deleted, affected individuals are less likely to develop hypertension. Therefore, the loss of this gene appears to be a protective factor. People with Williams syndrome whose NCF1 gene is not deleted have a higher risk of developing hypertension.
Several other genes are commonly part of the deletion on chromosome 7. Loss of some of these genes appears to be involved in particular signs and symptoms of the condition, and their relationship to the condition is under investigation. However, it is unknown what role, if any, the loss of many of these other genes plays in Williams syndrome.
Inheritance
Most cases of Williams syndrome are not inherited. The chromosomal alteration usually occurs as a random event during the formation of reproductive cells (eggs or sperm) in a parent of an affected individual. These cases occur in people with no history of the disorder in their family. However, the risk of having a child with Williams syndrome is increased if a parent, who is unaffected, has a chromosomal change called an inversion in the region of chromosome 7 associated with Williams syndrome.
Williams syndrome is considered an autosomal dominant condition because one copy of the altered chromosome 7 in each cell is sufficient to cause the disorder. In a small percentage of cases, people with Williams syndrome inherit the chromosomal deletion from a parent with the condition .
.
Other Names for This Condition
- Beuren syndrome
- Elfin facies syndrome
- Elfin facies with hypercalcemia
- Hypercalcemia-supravalvar aortic stenosis
- WBS
- Williams-Beuren syndrome
- WS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bhattacharjee Y. Friendly faces and unusual minds. Science. 2005 Nov 4;310(5749):802-4. doi: 10.1126/science.310.5749.802. No abstract available. Citation on PubMed
- Carrasco X, Castillo S, Aravena T, Rothhammer P, Aboitiz F. Williams syndrome: pediatric, neurologic, and cognitive development. Pediatr Neurol. 2005 Mar;32(3):166-72. doi: 10.1016/j.pediatrneurol.2004.09.013. Citation on PubMed
- Del Campo M, Antonell A, Magano LF, Munoz FJ, Flores R, Bayes M, Perez Jurado LA. Hemizygosity at the NCF1 gene in patients with Williams-Beuren syndrome decreases their risk of hypertension. Am J Hum Genet. 2006 Apr;78(4):533-42. doi: 10.1086/501073. Epub 2006 Jan 31. Citation on PubMed or Free article on PubMed Central
- Eckert MA, Galaburda AM, Mills DL, Bellugi U, Korenberg JR, Reiss AL. The neurobiology of Williams syndrome: cascading influences of visual system impairment? Cell Mol Life Sci. 2006 Aug;63(16):1867-75. doi: 10.1007/s00018-005-5553-x. Citation on PubMed
- Hobart HH, Morris CA, Mervis CB, Pani AM, Kistler DJ, Rios CM, Kimberley KW, Gregg RG, Bray-Ward P. Inversion of the Williams syndrome region is a common polymorphism found more frequently in parents of children with Williams syndrome. Am J Med Genet C Semin Med Genet. 2010 May 15;154C(2):220-8. doi: 10.1002/ajmg.c.30258. Citation on PubMed
- Kozel BA, Barak B, Kim CA, Mervis CB, Osborne LR, Porter M, Pober BR. Williams syndrome. Nat Rev Dis Primers. 2021 Jun 17;7(1):42. doi: 10.1038/s41572-021-00276-z. Citation on PubMed
- Kozel BA, Bayliss SJ, Berk DR, Waxler JL, Knutsen RH, Danback JR, Pober BR. Skin findings in Williams syndrome. Am J Med Genet A. 2014 Sep;164A(9):2217-25. doi: 10.1002/ajmg.a.36628. Epub 2014 Jun 11. Citation on PubMed
- Kozel BA, Danback JR, Waxler JL, Knutsen RH, de las Fuentes L, Reusz GS, Kis E, Bhatt AB, Pober BR. Williams syndrome predisposes to vascular stiffness modified by antihypertensive use and copy number changes in NCF1. Hypertension. 2014 Jan;63(1):74-9. doi: 10.1161/HYPERTENSIONAHA.113.02087. Epub 2013 Oct 14. Citation on PubMed
- Matisoff AJ, Olivieri L, Schwartz JM, Deutsch N. Risk assessment and anesthetic management of patients with Williams syndrome: a comprehensive review. Paediatr Anaesth. 2015 Dec;25(12):1207-15. doi: 10.1111/pan.12775. Epub 2015 Oct 12. Citation on PubMed
- Mervis CB, Becerra AM. Language and communicative development in Williams syndrome. Ment Retard Dev Disabil Res Rev. 2007;13(1):3-15. doi: 10.1002/mrdd.20140. Citation on PubMed
- Meyer-Lindenberg A, Hariri AR, Munoz KE, Mervis CB, Mattay VS, Morris CA, Berman KF. Neural correlates of genetically abnormal social cognition in Williams syndrome. Nat Neurosci. 2005 Aug;8(8):991-3. doi: 10.1038/nn1494. Epub 2005 Jul 10. Citation on PubMed
- Meyer-Lindenberg A, Mervis CB, Berman KF. Neural mechanisms in Williams syndrome: a unique window to genetic influences on cognition and behaviour. Nat Rev Neurosci. 2006 May;7(5):380-93. doi: 10.1038/nrn1906. Citation on PubMed
- Morris CA, Braddock SR; COUNCIL ON GENETICS. Health Care Supervision for Children With Williams Syndrome. Pediatrics. 2020 Feb;145(2):e20193761. doi: 10.1542/peds.2019-3761. Epub 2020 Jan 21. Citation on PubMed
- Morris CA. Williams Syndrome. 1999 Apr 9 [updated 2023 Apr 13]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1249/ Citation on PubMed
- Palacios-Verdu MG, Segura-Puimedon M, Borralleras C, Flores R, Del Campo M, Campuzano V, Perez-Jurado LA. Metabolic abnormalities in Williams-Beuren syndrome. J Med Genet. 2015 Apr;52(4):248-55. doi: 10.1136/jmedgenet-2014-102713. Epub 2015 Feb 6. Citation on PubMed
- Pober BR, Morris CA. Diagnosis and management of medical problems in adults with Williams-Beuren syndrome. Am J Med Genet C Semin Med Genet. 2007 Aug 15;145C(3):280-90. doi: 10.1002/ajmg.c.30139. Citation on PubMed
- Schubert C. The genomic basis of the Williams-Beuren syndrome. Cell Mol Life Sci. 2009 Apr;66(7):1178-97. doi: 10.1007/s00018-008-8401-y. Citation on PubMed
- Sindhar S, Lugo M, Levin MD, Danback JR, Brink BD, Yu E, Dietzen DJ, Clark AL, Purgert CA, Waxler JL, Elder RW, Pober BR, Kozel BA. Hypercalcemia in Patients with Williams-Beuren Syndrome. J Pediatr. 2016 Nov;178:254-260.e4. doi: 10.1016/j.jpeds.2016.08.027. Epub 2016 Aug 26. Citation on PubMed
- Stromme P, Bjornstad PG, Ramstad K. Prevalence estimation of Williams syndrome. J Child Neurol. 2002 Apr;17(4):269-71. doi: 10.1177/088307380201700406. Citation on PubMed
- Tassabehji M, Hammond P, Karmiloff-Smith A, Thompson P, Thorgeirsson SS, Durkin ME, Popescu NC, Hutton T, Metcalfe K, Rucka A, Stewart H, Read AP, Maconochie M, Donnai D. GTF2IRD1 in craniofacial development of humans and mice. Science. 2005 Nov 18;310(5751):1184-7. doi: 10.1126/science.1116142. Epub 2005 Nov 3. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.