

Description
Vici syndrome is a severe disorder that begins early in life and affects many body systems. It is characterized by abnormalities of the brain, immune system, heart, skin, and eyes. Other organs and tissues are less commonly affected.
A characteristic feature of Vici syndrome is a brain abnormality called agenesis of the corpus callosum, in which the tissue that connects the left and right halves of the brain (the corpus callosum ) fails to form normally during the early stages of development before birth. A region of the brain known as the pons
) fails to form normally during the early stages of development before birth. A region of the brain known as the pons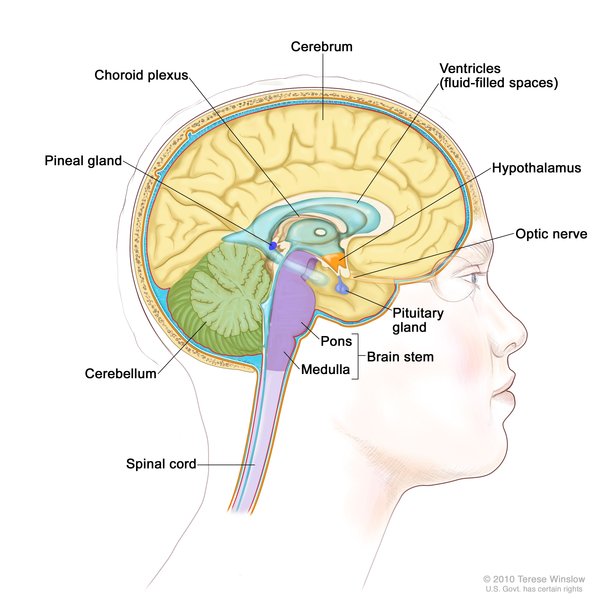 (pontine hypoplasia) may be underdeveloped in people with Vici syndrome. Affected individuals may also have lower levels of myelin
(pontine hypoplasia) may be underdeveloped in people with Vici syndrome. Affected individuals may also have lower levels of myelin , which is a fatty substance that covers and protects nerve cells. In addition to problems with brain development, breakdown (degeneration) of brain tissue may occur over time, resulting in an unusually small head size (microcephaly
, which is a fatty substance that covers and protects nerve cells. In addition to problems with brain development, breakdown (degeneration) of brain tissue may occur over time, resulting in an unusually small head size (microcephaly ).
).
These brain problems contribute to profound developmental delays in individuals with Vici syndrome. Affected infants have weak muscle tone (hypotonia). Generally, children with Vici syndrome are not able to roll or sit, and those that can may lose this skill when they get older. In addition, affected children cannot walk or speak.
Another characteristic feature of Vici syndrome is impaired immune function (immune deficiency), which leads to recurrent infections that can be life-threatening. Respiratory infections are the most common type of infection, though gastrointestinal and urinary tract infections also frequently occur.
A potentially life-threatening heart condition called cardiomyopathy is common in children with Vici syndrome. This condition, which worsens over time, makes it difficult for the heart to pump blood efficiently. Some affected children also have heart defects that are present from birth (congenital).
People with Vici syndrome may have skin and hair that are lighter in color than that of family members and other people with the same ethnic background (hypopigmentation). They may also experience clouding of the lenses of the eyes (cataracts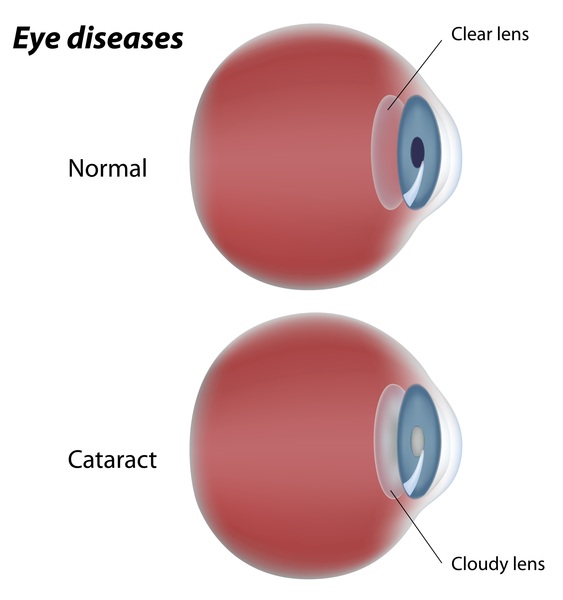 ) or other eye abnormalities, which may reduce their ability to see.
) or other eye abnormalities, which may reduce their ability to see.
Other, less common signs and symptoms of Vici syndrome include seizures; hearing loss caused by abnormalities of the inner ear (sensorineural hearing loss); an opening in the upper lip (cleft lip
(sensorineural hearing loss); an opening in the upper lip (cleft lip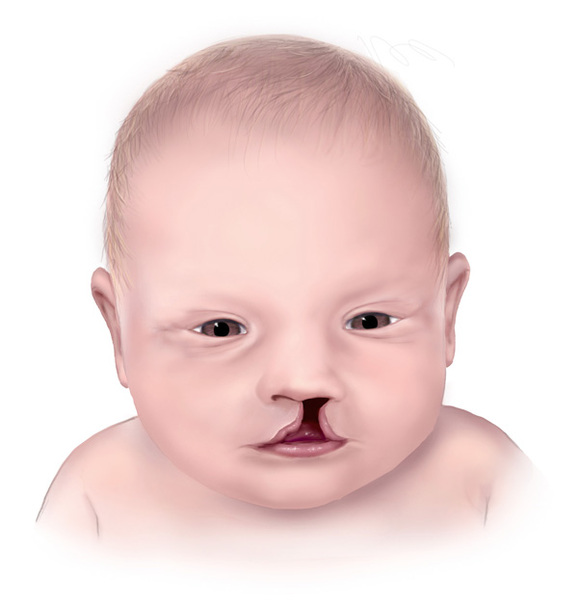 ) with or without an opening in the roof of the mouth (cleft palate
) with or without an opening in the roof of the mouth (cleft palate ) or other unusual facial features; and abnormal function of the thyroid, liver, or kidneys. Many affected infants grow and gain weight more slowly than expected.
) or other unusual facial features; and abnormal function of the thyroid, liver, or kidneys. Many affected infants grow and gain weight more slowly than expected.
Most people with Vici syndrome do not survive beyond childhood, though this can vary widely.
Frequency
Vici syndrome is a rare disorder, though its exact prevalence is unknown. Approximately 100 individuals have been diagnosed with this condition.
Causes
Variants (also called mutations) in the EPG5 gene cause Vici syndrome. This gene provides instructions for making a protein that is involved in a cellular process called autophagy. Cells use this process to recycle or break down worn-out or unnecessary cell parts. Autophagy also helps cells use materials efficiently when energy demands are high. In addition to its role in autophagy, the EPG5 protein aids in the body's immune response to foreign invaders such as bacteria and viruses.
Some variants in the EPG5 gene cause the gene to produce abnormal EPG5 proteins that do not respond properly to foreign invaders. This leads to recurrent infections. In addition, autophagy is impaired. Researchers believe that problems with autophagy can disrupt the normal development and survival of cells in the brain and other organs and tissues; however, they do not fully understand how this disruption leads to the signs and symptoms of Vici syndrome.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Absent corpus callosum cataract immunodeficiency
- Corpus callosum agenesis-cataract-immunodeficiency syndrome
- Dionisi Vici Sabetta Gambarara syndrome
- Dionisi-Vici-Sabetta-Gambarara syndrome
- Immunodeficiency with cleft lip/palate, cataract, hypopigmentation and absent corpus callosum
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Byrne S, Dionisi-Vici C, Smith L, Gautel M, Jungbluth H. Vici syndrome: a review. Orphanet J Rare Dis. 2016 Feb 29;11:21. doi: 10.1186/s13023-016-0399-x. Citation on PubMed or Free article on PubMed Central
- Byrne S, Jansen L, U-King-Im JM, Siddiqui A, Lidov HG, Bodi I, Smith L, Mein R, Cullup T, Dionisi-Vici C, Al-Gazali L, Al-Owain M, Bruwer Z, Al Thihli K, El-Garhy R, Flanigan KM, Manickam K, Zmuda E, Banks W, Gershoni-Baruch R, Mandel H, Dagan E, Raas-Rothschild A, Barash H, Filloux F, Creel D, Harris M, Hamosh A, Kolker S, Ebrahimi-Fakhari D, Hoffmann GF, Manchester D, Boyer PJ, Manzur AY, Lourenco CM, Pilz DT, Kamath A, Prabhakar P, Rao VK, Rogers RC, Ryan MM, Brown NJ, McLean CA, Said E, Schara U, Stein A, Sewry C, Travan L, Wijburg FA, Zenker M, Mohammed S, Fanto M, Gautel M, Jungbluth H. EPG5-related Vici syndrome: a paradigm of neurodevelopmental disorders with defective autophagy. Brain. 2016 Mar;139(Pt 3):765-81. doi: 10.1093/brain/awv393. Citation on PubMed or Free article on PubMed Central
- Cullup T, Kho AL, Dionisi-Vici C, Brandmeier B, Smith F, Urry Z, Simpson MA, Yau S, Bertini E, McClelland V, Al-Owain M, Koelker S, Koerner C, Hoffmann GF, Wijburg FA, ten Hoedt AE, Rogers RC, Manchester D, Miyata R, Hayashi M, Said E, Soler D, Kroisel PM, Windpassinger C, Filloux FM, Al-Kaabi S, Hertecant J, Del Campo M, Buk S, Bodi I, Goebel HH, Sewry CA, Abbs S, Mohammed S, Josifova D, Gautel M, Jungbluth H. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet. 2013 Jan;45(1):83-7. doi: 10.1038/ng.2497. Epub 2012 Dec 9. Citation on PubMed or Free article on PubMed Central
- Hori I, Otomo T, Nakashima M, Miya F, Negishi Y, Shiraishi H, Nonoda Y, Magara S, Tohyama J, Okamoto N, Kumagai T, Shimoda K, Yukitake Y, Kajikawa D, Morio T, Hattori A, Nakagawa M, Ando N, Nishino I, Kato M, Tsunoda T, Saitsu H, Kanemura Y, Yamasaki M, Kosaki K, Matsumoto N, Yoshimori T, Saitoh S. Defects in autophagosome-lysosome fusion underlie Vici syndrome, a neurodevelopmental disorder with multisystem involvement. Sci Rep. 2017 Jun 14;7(1):3552. doi: 10.1038/s41598-017-02840-8. Citation on PubMed or Free article on PubMed Central
- Piano Mortari E, Folgiero V, Marcellini V, Romania P, Bellacchio E, D'Alicandro V, Bocci C, Carrozzo R, Martinelli D, Petrini S, Axiotis E, Farroni C, Locatelli F, Schara U, Pilz DT, Jungbluth H, Dionisi-Vici C, Carsetti R. The Vici syndrome protein EPG5 regulates intracellular nucleic acid trafficking linking autophagy to innate and adaptive immunity. Autophagy. 2018;14(1):22-37. doi: 10.1080/15548627.2017.1389356. Epub 2018 Jan 2. Citation on PubMed or Free article on PubMed Central
- Vansenne F, Fock JM, Stolte-Dijkstra I, Meiners LC, van den Boogaard MH, Jaeger B, Boven L, Vos YJ, Sinke RJ, Verbeek DS. Phenotypic expansion of EGP5-related Vici syndrome: 15 Dutch patients carrying a founder variant. Eur J Paediatr Neurol. 2022 Nov;41:91-98. doi: 10.1016/j.ejpn.2022.11.003. Epub 2022 Nov 12. Citation on PubMed
- Wang Z, Miao G, Xue X, Guo X, Yuan C, Wang Z, Zhang G, Chen Y, Feng D, Hu J, Zhang H. The Vici Syndrome Protein EPG5 Is a Rab7 Effector that Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes. Mol Cell. 2016 Sep 1;63(5):781-95. doi: 10.1016/j.molcel.2016.08.021. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.