

Description
Oral-facial-digital syndrome is actually a group of related conditions that affect the development of the oral cavity (the mouth and teeth), facial features, and digits (fingers and toes).
Researchers have identified at least 13 potential forms of oral-facial-digital syndrome. The different types are classified by their patterns of signs and symptoms. However, the features of the various types overlap significantly, and some types are not well defined. The classification system for oral-facial-digital syndrome continues to evolve as researchers find more affected individuals and learn more about this disorder.
The signs and symptoms of oral-facial-digital syndrome vary widely. However, most forms of this disorder involve problems with development of the oral cavity, facial features, and digits. Most forms are also associated with brain abnormalities and some degree of intellectual disability.
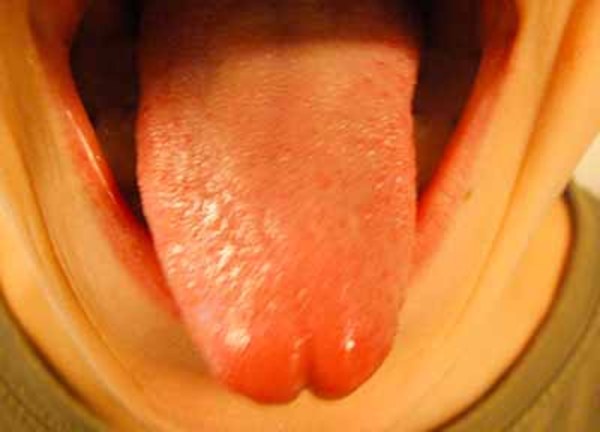
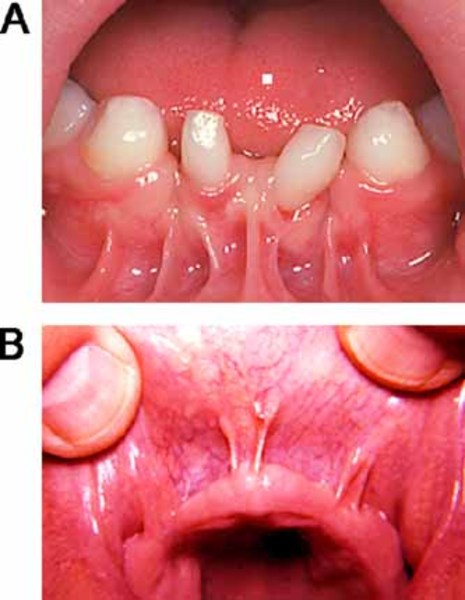
Abnormalities of the oral cavity that occur in many types of oral-facial-digital syndrome include a split (cleft) in the tongue, a tongue with an unusual lobed shape, and the growth of noncancerous tumors or nodules on the tongue. Affected individuals may also have extra, missing, or defective teeth. Another common feature is an opening in the roof of the mouth (a cleft palate). Some people with oral-facial-digital syndrome have bands of extra tissue (called hyperplastic frenula) that abnormally attach the lip to the gums.


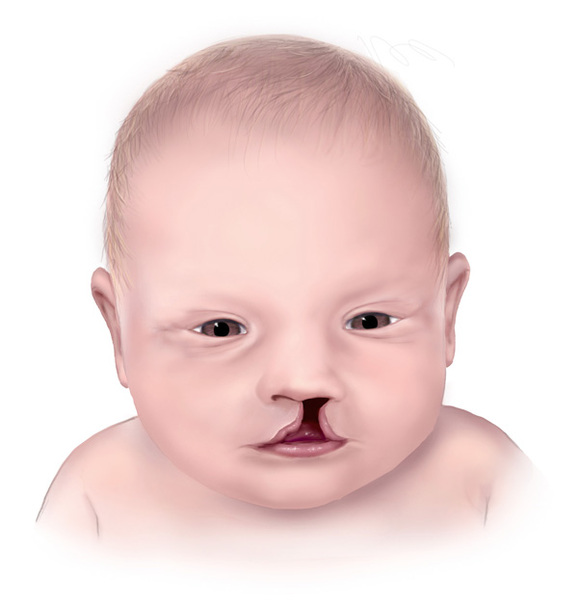
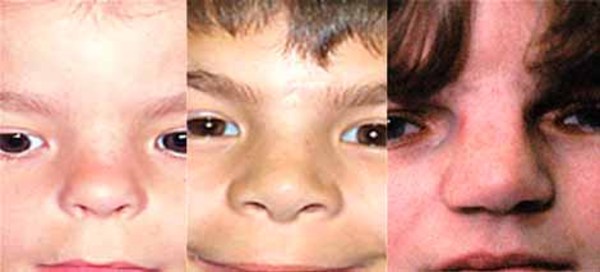
Distinctive facial features often associated with oral-facial-digital syndrome include a split in the lip (a cleft lip); a wide nose with a broad, flat nasal bridge; and widely spaced eyes (hypertelorism).

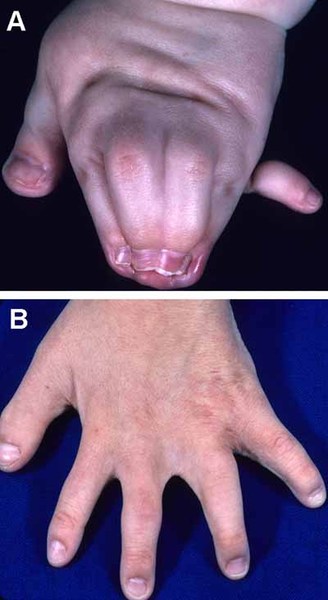
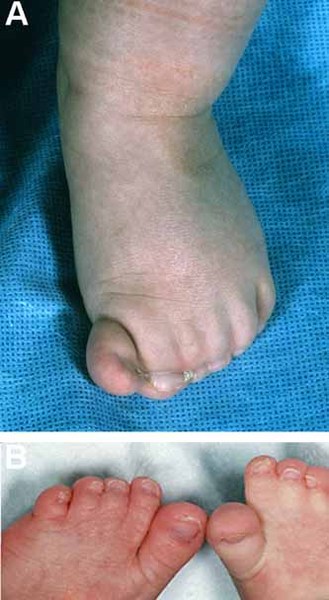
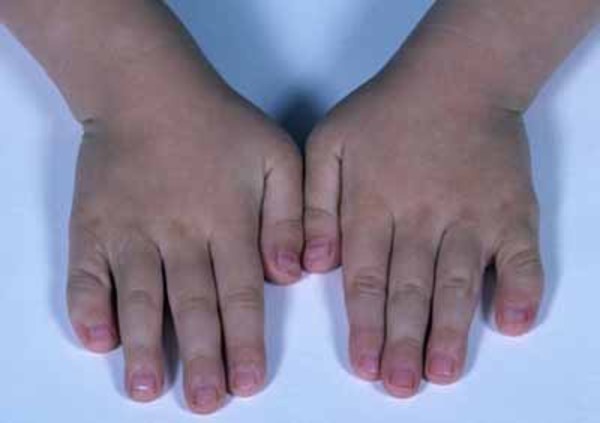
Abnormalities of the digits can affect both the fingers and the toes in people with oral-facial-digital syndrome. These abnormalities include fusion of certain fingers or toes (syndactyly), digits that are shorter than usual (brachydactyly), or digits that are unusually curved (clinodactyly). The presence of extra digits (polydactyly) is also seen in most forms of oral-facial-digital syndrome.

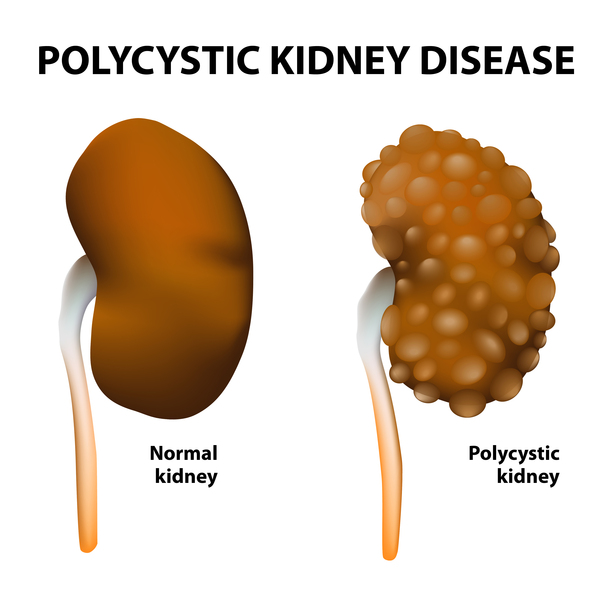
Other features occur in only one or a few types of oral-facial digital syndrome. These features help distinguish the different forms of the disorder. For example, the most common form of oral-facial-digital syndrome, type I, is associated with polycystic kidney disease. This kidney disease is characterized by the growth of fluid-filled sacs (cysts) that interfere with the kidneys' ability to filter waste products from the blood. Other forms of oral-facial-digital syndrome are characterized by neurological problems, particular changes in the structure of the brain, bone abnormalities, vision loss, and heart defects.
Frequency
Oral-facial-digital syndrome has an estimated incidence of 1 in 50,000 to 250,000 newborns. Type I accounts for the majority of cases of this disorder. The other forms of oral-facial-digital syndrome are very rare; most have been identified in only one or a few families.
Causes
Only one gene, OFD1, has been associated with oral-facial-digital syndrome. Mutations in this gene cause oral-facial-digital syndrome type I. OFD1 gene mutations were also found in an affected family whose disorder was classified as type VII; however, researchers now believe that type VII is the same as type I.
The OFD1 gene provides instructions for making a protein whose function is not fully understood. It appears to play an important role in the early development of many parts of the body, including the brain, face, limbs, and kidneys. Mutations in the OFD1 gene prevent cells from making enough functional OFD1 protein, which disrupts the normal development of these structures. It is unclear how a shortage of this protein causes the specific features of oral-facial-digital syndrome type I.
Researchers are actively searching for the genetic changes responsible for the other forms of oral-facial-digital syndrome.
Inheritance
Oral-facial-digital syndrome type I is inherited in an X-linked dominant pattern. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the gene in each cell is sufficient to cause the disorder. Some cells produce a normal amount of OFD1 protein and other cells produce none. The resulting overall reduction in the amount of this protein leads to the signs and symptoms of oral-facial-digital syndrome type I.
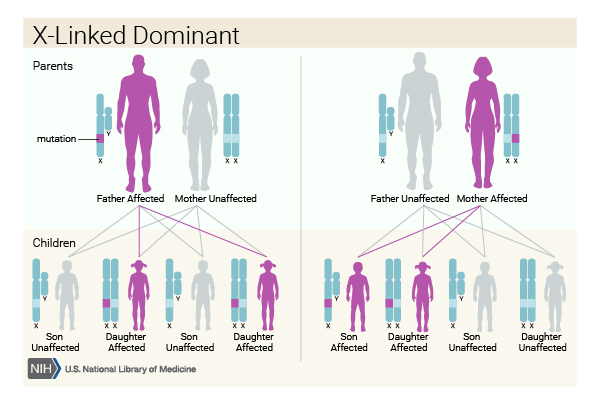

In males (who have only one X chromosome), mutations result in a total loss of the OFD1 protein. A lack of this protein is usually lethal very early in development, so very few males are born with oral-facial-digital syndrome type I. Affected males usually die before birth, although a few have lived into early infancy.
Most of the other forms of oral-facial-digital syndrome are inherited in an autosomal recessive pattern, which suggests that both copies of a causative gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Dysplasia linguofacialis
- OFDS
- Oro-facio-digital syndrome
- Orodigitofacial dysostosis
- Orodigitofacial syndrome
- Orofaciodigital syndrome
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Orofacial-digital syndrome III

- Genetic Testing Registry: Orofacial-digital syndrome IV
- Genetic Testing Registry: Orofaciodigital syndrome
- Genetic Testing Registry: Orofaciodigital syndrome I
- Genetic Testing Registry: Orofaciodigital syndrome type 6
- Genetic Testing Registry: Orofaciodigital syndrome V
- Genetic Testing Registry: Orofaciodigital syndrome VIII
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- OROFACIODIGITAL SYNDROME X; OFD10
- OROFACIODIGITAL SYNDROME VI; OFD6
- OROFACIODIGITAL SYNDROME II; OFD2
- OROFACIODIGITAL SYNDROME VIII; OFD8
- OROFACIODIGITAL SYNDROME I; OFD1
- OROFACIODIGITAL SYNDROME III; OFD3
- OROFACIODIGITAL SYNDROME IV; OFD4
- OROFACIODIGITAL SYNDROME IX; OFD9
- OROFACIODIGITAL SYNDROME V; OFD5
- OROFACIODIGITAL SYNDROME VII; OFD7
- OROFACIODIGITAL SYNDROME XI; OFD11
Scientific Articles on PubMed
References
- Ferrante MI, Giorgio G, Feather SA, Bulfone A, Wright V, Ghiani M, Selicorni A, Gammaro L, Scolari F, Woolf AS, Sylvie O, Bernard L, Malcolm S, Winter R, Ballabio A, Franco B. Identification of the gene for oral-facial-digital type I syndrome. Am J Hum Genet. 2001 Mar;68(3):569-76. doi: 10.1086/318802. Epub 2001 Feb 13. Citation on PubMed or Free article on PubMed Central
- Gurrieri F, Franco B, Toriello H, Neri G. Oral-facial-digital syndromes: review and diagnostic guidelines. Am J Med Genet A. 2007 Dec 15;143A(24):3314-23. doi: 10.1002/ajmg.a.32032. Citation on PubMed
- Macca M, Franco B. The molecular basis of oral-facial-digital syndrome, type 1. Am J Med Genet C Semin Med Genet. 2009 Nov 15;151C(4):318-25. doi: 10.1002/ajmg.c.30224. Citation on PubMed
- Nowaczyk MJ, Zeesman S, Whelan DT, Wright V, Feather SA. Oral-facial-digital syndrome VII is oral-facial-digital syndrome I: a clarification. Am J Med Genet A. 2003 Dec 1;123A(2):179-82. doi: 10.1002/ajmg.a.20215. Citation on PubMed
- Prattichizzo C, Macca M, Novelli V, Giorgio G, Barra A, Franco B; Oral-Facial-Digital Type I (OFDI) Collaborative Group. Mutational spectrum of the oral-facial-digital type I syndrome: a study on a large collection of patients. Hum Mutat. 2008 Oct;29(10):1237-46. doi: 10.1002/humu.20792. Citation on PubMed
- Saal S, Faivre L, Aral B, Gigot N, Toutain A, Van Maldergem L, Destree A, Maystadt I, Cosyns JP, Jouk PS, Loeys B, Chauveau D, Bieth E, Layet V, Mathieu M, Lespinasse J, Teebi A, Franco B, Gautier E, Binquet C, Masurel-Paulet A, Mousson C, Gouyon JB, Huet F, Thauvin-Robinet C. Renal insufficiency, a frequent complication with age in oral-facial-digital syndrome type I. Clin Genet. 2010 Mar;77(3):258-65. doi: 10.1111/j.1399-0004.2009.01290.x. Epub 2009 Oct 8. Citation on PubMed
- Siebert JR. The oral-facial-digital syndromes. Handb Clin Neurol. 2008;87:341-51. doi: 10.1016/S0072-9752(07)87018-7. No abstract available. Citation on PubMed
- Thauvin-Robinet C, Cossee M, Cormier-Daire V, Van Maldergem L, Toutain A, Alembik Y, Bieth E, Layet V, Parent P, David A, Goldenberg A, Mortier G, Heron D, Sagot P, Bouvier AM, Huet F, Cusin V, Donzel A, Devys D, Teyssier JR, Faivre L. Clinical, molecular, and genotype-phenotype correlation studies from 25 cases of oral-facial-digital syndrome type 1: a French and Belgian collaborative study. J Med Genet. 2006 Jan;43(1):54-61. doi: 10.1136/jmg.2004.027672. Citation on PubMed or Free article on PubMed Central
- Toriello HV. Are the oral-facial-digital syndromes ciliopathies? Am J Med Genet A. 2009 May;149A(5):1089-95. doi: 10.1002/ajmg.a.32799. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.