

Description
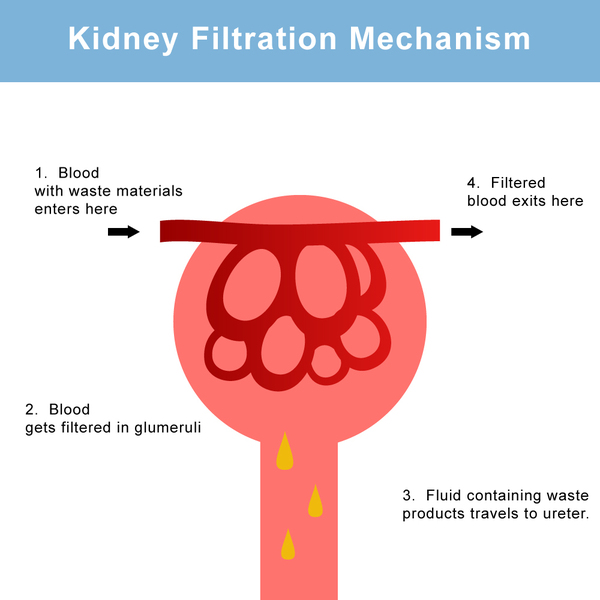
Polycystic kidney disease is a disorder that affects the kidneys and other organs. Clusters of fluid-filled sacs, called cysts, develop in the kidneys and interfere with their ability to filter waste products from the blood. The growth of cysts causes the kidneys to become enlarged and can lead to kidney failure. Cysts may also develop in other organs, particularly the liver.
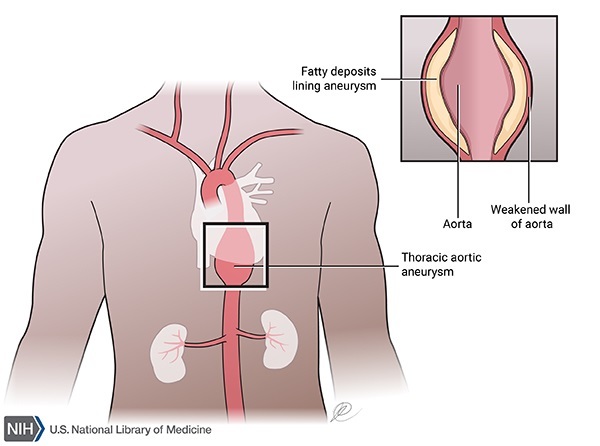
Frequent complications of polycystic kidney disease include dangerously high blood pressure (hypertension), pain in the back or sides, blood in the urine (hematuria), recurrent urinary tract infections, kidney stones, and heart valve abnormalities. Additionally, people with polycystic kidney disease have an increased risk of an abnormal bulging (an aneurysm) in a large blood vessel called the aorta or in blood vessels at the base of the brain. Aneurysms can be life-threatening if they tear or rupture.

The two major forms of polycystic kidney disease are distinguished by the usual age of onset and the pattern in which it is passed through families. The autosomal dominant form (sometimes called ADPKD) has signs and symptoms that typically begin in adulthood, although cysts in the kidney are often present from birth or childhood. Autosomal dominant polycystic kidney disease can be further divided into type 1 and type 2, depending on the genetic cause. The autosomal recessive form of polycystic kidney disease (sometimes called ARPKD) is much rarer and is often lethal early in life. The signs and symptoms of this condition are usually apparent at birth or in early infancy.
Frequency
Polycystic kidney disease is a fairly common genetic disorder. It affects about 500,000 people in the United States. The autosomal dominant form of the disease is much more common than the autosomal recessive form. Autosomal dominant polycystic kidney disease affects 1 in 500 to 1,000 people, while the autosomal recessive type occurs in an estimated 1 in 20,000 to 40,000 people.
Causes
Mutations in the PKD1, PKD2, and PKHD1 genes cause polycystic kidney disease.
Mutations in either the PKD1 or PKD2 gene can cause autosomal dominant polycystic kidney disease; PKD1 gene mutations cause ADPKD type 1, and PKD2 gene mutations cause ADPKD type 2. These genes provide instructions for making proteins whose functions are not fully understood. Researchers believe that they are involved in transmitting chemical signals from outside the cell to the cell's nucleus. The two proteins work together to promote normal kidney development, organization, and function. Mutations in the PKD1 or PKD2 gene lead to the formation of thousands of cysts, which disrupt the normal functions of the kidneys and other organs. People with mutations in the PKD2 gene, particularly women, typically have a less severe form of the disease than people with PKD1 mutations. The signs and symptoms, including a decline in kidney function, tend to appear later in adulthood in people with a PKD2 mutation.
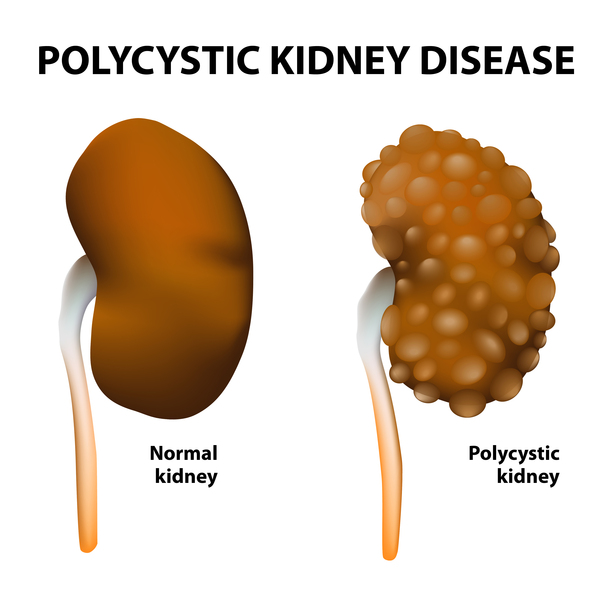
Mutations in the PKHD1 gene cause autosomal recessive polycystic kidney disease. This gene provides instructions for making a protein whose exact function is unknown; however, the protein likely transmits chemical signals from outside the cell to the cell nucleus. Researchers have not determined how mutations in the PKHD1 gene lead to the formation of numerous cysts characteristic of polycystic kidney disease.
Although polycystic kidney disease is usually a genetic disorder, a small percentage of cases are not caused by gene mutations. These cases are called acquired polycystic kidney disease. This form of the disorder occurs most often in people with other types of kidney disease who have been treated for several years with hemodialysis (a procedure that filters waste products from the blood).
Inheritance
Most cases of polycystic kidney disease have an autosomal dominant pattern of inheritance. People with this condition are born with one mutated copy of the PKD1 or PKD2 gene in each cell. In about 90 percent of these cases, an affected person inherits the mutation from one affected parent. The other 10 percent of cases result from a new mutation in one of the genes and occur in people with no history of the disorder in their family.


Although one altered copy of a gene in each cell is sufficient to cause the disorder, an additional mutation in the second copy of the PKD1 or PKD2 gene may make cysts grow faster and increase the severity of the disease. The rate at which cysts enlarge and cause a loss of kidney function varies widely, and may be influenced by mutations in other genes that have not been identified.
Polycystic kidney disease also can be inherited in an autosomal recessive pattern. People with this form of the condition have two altered copies of the PKHD1 gene in each cell. The parents of a child with an autosomal recessive disorder are not affected but are carriers of one copy of the altered gene.

Other Names for This Condition
- PKD
- Polycystic renal disease
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Autosomal recessive polycystic kidney disease

- Genetic Testing Registry: Autosomal dominant polycystic kidney disease
- Genetic Testing Registry: Polycystic kidney disease 3 with or without polycystic liver disease
- Genetic Testing Registry: Polycystic kidney disease 2
- Genetic Testing Registry: Polycystic kidney disease, adult type
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Adeva M, El-Youssef M, Rossetti S, Kamath PS, Kubly V, Consugar MB, Milliner DM, King BF, Torres VE, Harris PC. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine (Baltimore). 2006 Jan;85(1):1-21. doi: 10.1097/01.md.0000200165.90373.9a. Citation on PubMed
- Boucher C, Sandford R. Autosomal dominant polycystic kidney disease (ADPKD, MIM 173900, PKD1 and PKD2 genes, protein products known as polycystin-1 and polycystin-2). Eur J Hum Genet. 2004 May;12(5):347-54. doi: 10.1038/sj.ejhg.5201162. Citation on PubMed
- Burgmaier K, Gimpel C, Schaefer F, Liebau M. Autosomal Recessive Polycystic Kidney Disease - PKHD1. 2001 Jul 19 [updated 2024 Apr 4]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1326/ Citation on PubMed
- Eccles MR, Stayner CA. Polycystic kidney disease - where gene dosage counts. F1000Prime Rep. 2014 Apr 1;6:24. doi: 10.12703/P6-24. eCollection 2014. Citation on PubMed or Free article on PubMed Central
- Harris PC, Torres VE. Polycystic Kidney Disease, Autosomal Dominant. 2002 Jan 10 [updated 2022 Sep 29]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1246/ Citation on PubMed
- Hateboer N, v Dijk MA, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, Torra R, Breuning M, Ravine D. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet. 1999 Jan 9;353(9147):103-7. doi: 10.1016/s0140-6736(98)03495-3. Citation on PubMed
- Horie S. ADPKD: molecular characterization and quest for treatment. Clin Exp Nephrol. 2005 Dec;9(4):282-291. doi: 10.1007/s10157-005-0367-6. Citation on PubMed
- Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002 Sep;13(9):2384-98. doi: 10.1097/01.asn.0000028643.17901.42. No abstract available. Citation on PubMed
- Lina F, Satlinb LM. Polycystic kidney disease: the cilium as a common pathway in cystogenesis. Curr Opin Pediatr. 2004 Apr;16(2):171-6. doi: 10.1097/00008480-200404000-00010. Citation on PubMed
- Sweeney WE Jr, Avner ED. Pathophysiology of childhood polycystic kidney diseases: new insights into disease-specific therapy. Pediatr Res. 2014 Jan;75(1-2):148-57. doi: 10.1038/pr.2013.191. Epub 2013 Oct 31. Citation on PubMed or Free article on PubMed Central
- Tahvanainen E, Tahvanainen P, Kaariainen H, Hockerstedt K. Polycystic liver and kidney diseases. Ann Med. 2005;37(8):546-55. doi: 10.1080/07853890500389181. Citation on PubMed
- Wilson PD. Polycystic kidney disease. N Engl J Med. 2004 Jan 8;350(2):151-64. doi: 10.1056/NEJMra022161. No abstract available. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.