

Description
Nonsyndromic holoprosencephaly is an abnormality of brain development that also affects the head and face. Normally, the brain divides into two halves (hemispheres) during early development. Holoprosencephaly occurs when the brain fails to divide properly into the right and left hemispheres. This condition is called nonsyndromic to distinguish it from other types of holoprosencephaly caused by genetic syndromes, chromosome abnormalities, or substances that cause birth defects (teratogens). The severity of nonsyndromic holoprosencephaly varies widely among affected individuals, even within the same family.

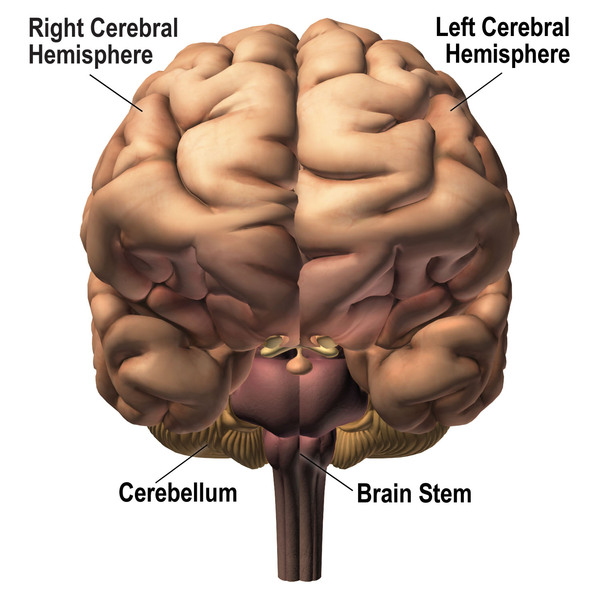
Nonsyndromic holoprosencephaly can be grouped into four types according to the degree of brain division. From most to least severe, the types are known as alobar, semi-lobar, lobar, and middle interhemispheric variant (MIHV). In the most severe forms of nonsyndromic holoprosencephaly, the brain does not divide at all. These affected individuals have one central eye (cyclopia) and a tubular nasal structure (proboscis) located above the eye. Most babies with severe nonsyndromic holoprosencephaly die before birth or soon after. In the less severe forms, the brain is partially divided and the eyes are usually set close together (hypotelorism). The life expectancy of these affected individuals varies depending on the severity of symptoms.
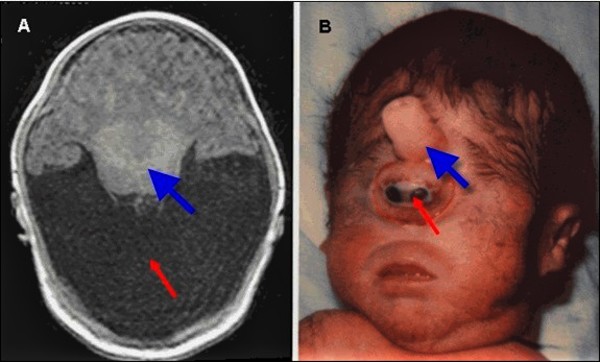
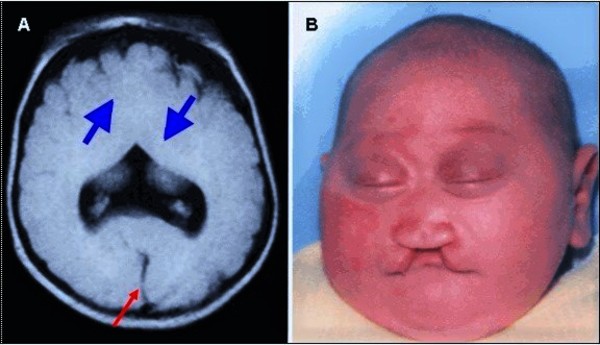
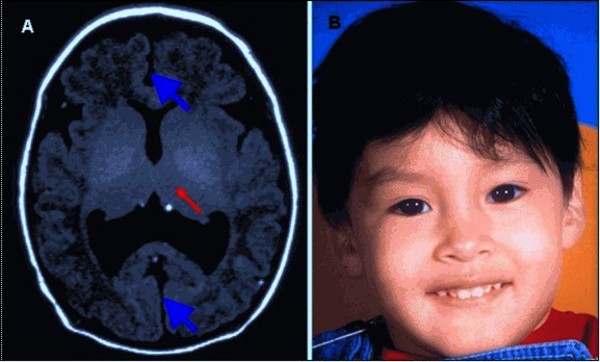
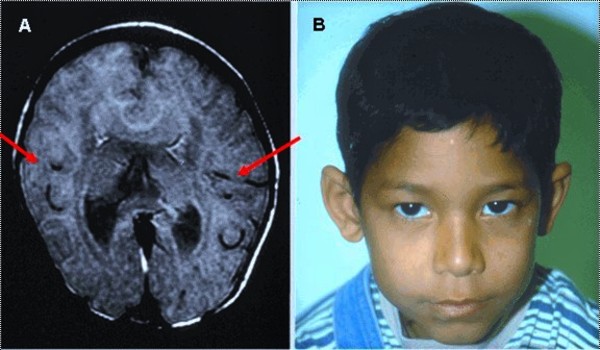
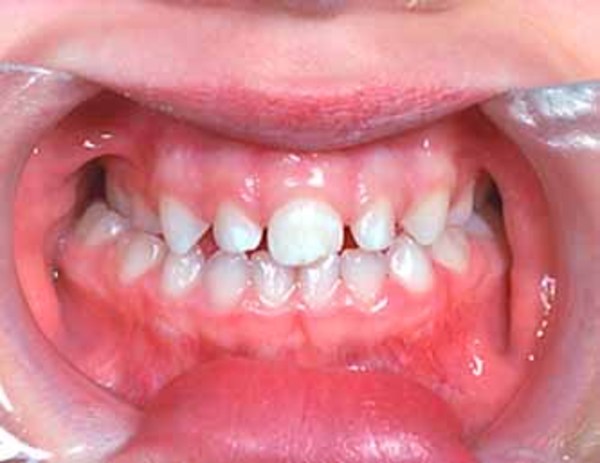
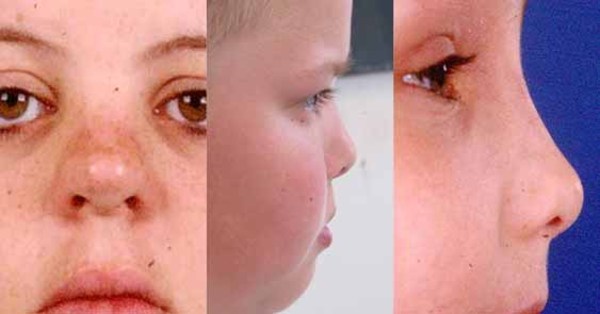
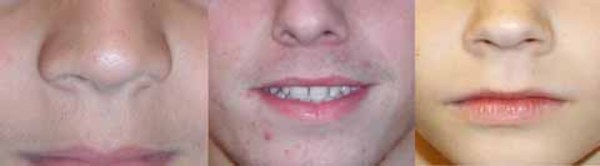
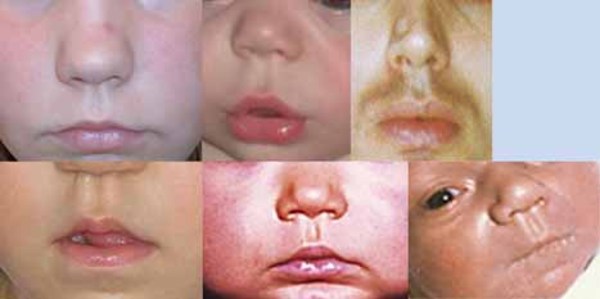
People with nonsyndromic holoprosencephaly often have a small head (microcephaly), although they can develop a buildup of fluid in the brain (hydrocephalus) that causes increased head size (macrocephaly). Other features may include an opening in the roof of the mouth (cleft palate) with or without a split in the upper lip (cleft lip), one central front tooth instead of two (a single maxillary central incisor), and a flat nasal bridge. The eyeballs may be abnormally small (microphthalmia) or absent (anophthalmia).

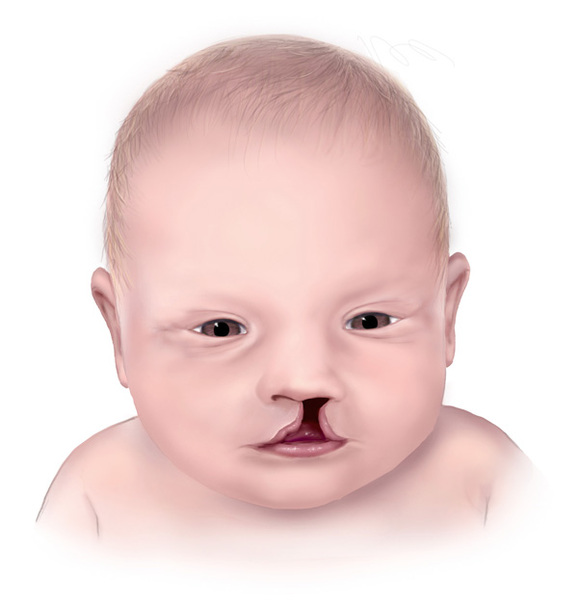
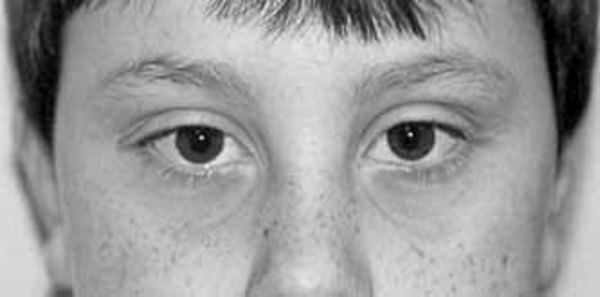
Some individuals with nonsyndromic holoprosencephaly have a distinctive pattern of facial features, including a narrowing of the head at the temples, outside corners of the eyes that point upward (upslanting palpebral fissures), large ears, a short nose with upturned nostrils, and a broad and deep space between the nose and mouth (philtrum). In general, the severity of facial features is directly related to the severity of the brain abnormalities. However, individuals with mildly affected facial features can have severe brain abnormalities. Some people do not have apparent structural brain abnormalities but have some of the facial features associated with this condition. These individuals are considered to have a form of the disorder known as microform holoprosencephaly and are typically identified after the birth of a severely affected family member.
Most people with nonsyndromic holoprosencephaly have developmental delay and intellectual disability. Affected individuals also frequently have a malfunctioning pituitary gland, which is a gland located at the base of the brain that produces several hormones. Because pituitary dysfunction leads to the partial or complete absence of these hormones, it can cause a variety of disorders. Most commonly, people with nonsyndromic holoprosencephaly and pituitary dysfunction develop diabetes insipidus, a condition that disrupts the balance between fluid intake and urine excretion. Dysfunction in other parts of the brain can cause seizures, feeding difficulties, and problems regulating body temperature, heart rate, and breathing. The sense of smell may be diminished (hyposmia) or completely absent (anosmia) if the part of the brain that processes smells is underdeveloped or missing.
Frequency
Nonsyndromic holoprosencephaly accounts for approximately 25 to 50 percent of all cases of holoprosencephaly, which affects an estimated 1 in 10,000 newborns.
Causes
Mutations in 11 genes have been found to cause nonsyndromic holoprosencephaly. These genes provide instructions for making proteins that are important for normal embryonic development, particularly for determining the shape of the brain and face. About 25 percent of people with nonsyndromic holoprosencephaly have a mutation in one of these four genes: SHH, ZIC2, SIX3, or TGIF1. Mutations in the other genes related to nonsyndromic holoprosencephaly are found in only a small percentage of cases. Many individuals with this condition do not have an identified gene mutation. The cause of the disorder is unknown in these individuals.
The brain normally divides into right and left hemispheres during the third to fourth week of pregnancy. To establish the line that separates the two hemispheres (the midline), the activity of many genes must be tightly regulated and coordinated. These genes provide instructions for making signaling proteins, which instruct the cells within the brain to form the right and left hemispheres.
Signaling proteins are also important for the formation of the eyes. During early development, the cells that develop into the eyes form a single structure called the eye field. This structure is located in the center of the developing face. The signaling protein produced from the SHH gene causes the eye field to separate into two distinct eyes. The SIX3 gene is involved in the formation of the lens of the eye and the specialized tissue at the back of the eye that detects light and color (the retina).

Mutations in the genes that cause nonsyndromic holoprosencephaly lead to the production of abnormal or nonfunctional signaling proteins. Without the correct signals, the eyes will not form normally and the brain does not separate into two hemispheres. The development of other parts of the face is affected if the eyes do not move to their proper position. The signs and symptoms of nonsyndromic holoprosencephaly are caused by abnormal development of the brain and face.
Researchers believe that other genetic or environmental factors, many of which have not been identified, play a role in determining the severity of nonsyndromic holoprosencephaly.
Inheritance
Nonsyndromic holoprosencephaly is inherited in an autosomal dominant pattern, which means an alteration in one copy of a gene in each cell is usually sufficient to cause the disorder. However, not all people with a gene mutation will develop signs and symptoms of the condition.
In some cases, an affected person inherits the mutation from one parent who may or may not have mild features of the condition. Other cases result from a new gene mutation and occur in people with no history of the disorder in their family.


Other Names for This Condition
- Holoprosencephaly sequence
- Isolated holoprosencephaly
- Isolated HPE
- Non-syndromic, non-chromosomal holoprosencephaly
- Non-syndromic, non-chromosomal HPE
- Nonsyndromic HPE
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Scientific Articles on PubMed
References
- Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencephaly. Orphanet J Rare Dis. 2007 Feb 2;2:8. doi: 10.1186/1750-1172-2-8. Citation on PubMed or Free article on PubMed Central
- Fernandes M, Hebert JM. The ups and downs of holoprosencephaly: dorsal versus ventral patterning forces. Clin Genet. 2008 May;73(5):413-23. doi: 10.1111/j.1399-0004.2008.00994.x. Epub 2008 Apr 2. Citation on PubMed
- Monuki ES. The morphogen signaling network in forebrain development and holoprosencephaly. J Neuropathol Exp Neurol. 2007 Jul;66(7):566-75. doi: 10.1097/nen.0b013e3180986e1b. Citation on PubMed
- Orioli IM, Castilla EE. Epidemiology of holoprosencephaly: Prevalence and risk factors. Am J Med Genet C Semin Med Genet. 2010 Feb 15;154C(1):13-21. doi: 10.1002/ajmg.c.30233. Citation on PubMed
- Pineda-Alvarez DE, Dubourg C, David V, Roessler E, Muenke M. Current recommendations for the molecular evaluation of newly diagnosed holoprosencephaly patients. Am J Med Genet C Semin Med Genet. 2010 Feb 15;154C(1):93-101. doi: 10.1002/ajmg.c.30253. Citation on PubMed or Free article on PubMed Central
- Roessler E, Muenke M. The molecular genetics of holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010 Feb 15;154C(1):52-61. doi: 10.1002/ajmg.c.30236. Citation on PubMed or Free article on PubMed Central
- Solomon BD, Mercier S, Velez JI, Pineda-Alvarez DE, Wyllie A, Zhou N, Dubourg C, David V, Odent S, Roessler E, Muenke M. Analysis of genotype-phenotype correlations in human holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010 Feb 15;154C(1):133-41. doi: 10.1002/ajmg.c.30240. Citation on PubMed or Free article on PubMed Central
- Solomon BD, Pineda-Alvarez DE, Mercier S, Raam MS, Odent S, Muenke M. Holoprosencephaly flashcards: A summary for the clinician. Am J Med Genet C Semin Med Genet. 2010 Feb 15;154C(1):3-7. doi: 10.1002/ajmg.c.30245. Citation on PubMed
- Tekendo-Ngongang C, Muenke M, Kruszka P. Holoprosencephaly Overview. 2000 Dec 27 [updated 2020 Mar 5]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1530/ Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.