

Description
Li-Fraumeni syndrome is a rare disorder that greatly increases the risk of developing several types of cancer, particularly in children and young adults.
The cancers most often associated with Li-Fraumeni syndrome include breast cancer, a form of bone cancer called osteosarcoma, and cancers of soft tissues (such as muscle) called soft tissue sarcomas. Other cancers commonly seen in this syndrome include brain tumors, cancers of blood-forming tissues (leukemias), and a cancer called adrenocortical carcinoma that affects the outer layer of the adrenal glands (small hormone-producing glands on top of each kidney). Several other types of cancer also occur more frequently in people with Li-Fraumeni syndrome.
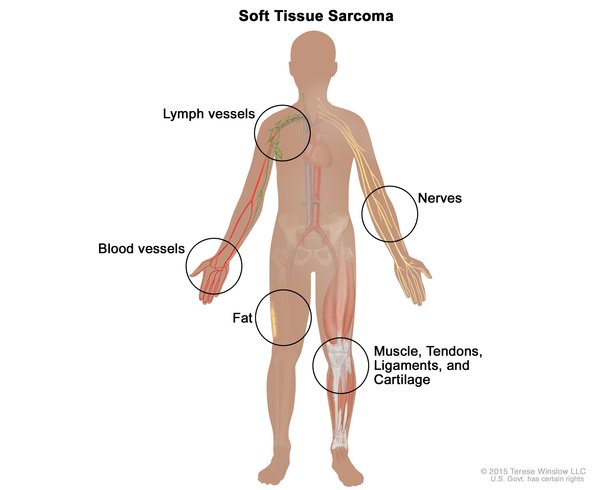
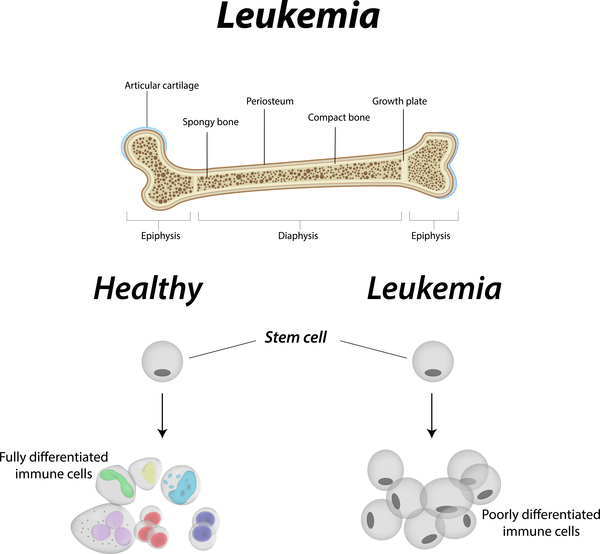
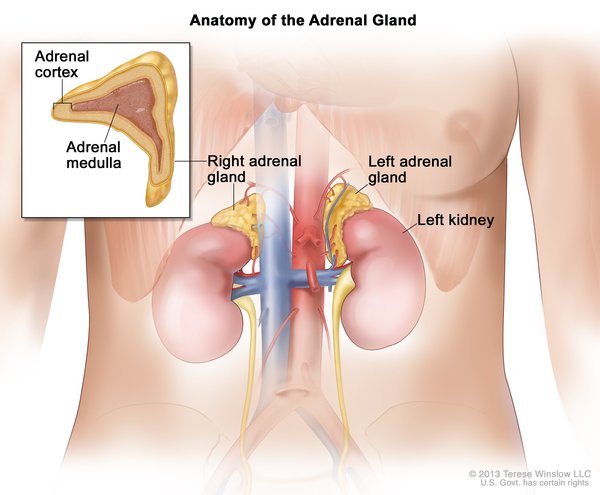
A very similar condition called Li-Fraumeni-like syndrome shares many of the features of classic Li-Fraumeni syndrome. Both conditions significantly increase the chances of developing multiple cancers beginning in childhood; however, the pattern of specific cancers seen in affected family members is different.
Frequency
Li-Fraumeni syndrome is thought to occur in 1 in 5,000 to 1 in 20,000 people worldwide.
Causes
Li-Fraumeni syndrome is associated with mutations in the TP53 gene. Nearly three-quarters of families with Li-Fraumeni syndrome and about one-quarter with Li-Fraumeni-like syndrome have germline mutations in the TP53 gene. Germline mutations are typically inherited and are present in essentially every cell in the body. TP53 is a tumor suppressor gene, which means that it normally helps control the growth and division of cells. Mutations in this gene can allow cells to divide in an uncontrolled way and form tumors. Other genetic and environmental factors are also likely to affect the risk of cancer in people with TP53 mutations.
A few families with cancers characteristic of Li-Fraumeni syndrome and Li-Fraumeni-like syndrome do not have TP53 mutations. The genetic factors involved in these cases are unclear.
Inheritance
Li-Fraumeni syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to increase the risk of developing cancer. Most people with Li-Fraumeni syndrome inherit an altered copy of the gene from an affected parent. In 7 to 20 percent of cases, however, the altered gene is the result of a new (de novo) mutation in the gene that occurred during the formation of reproductive cells (eggs or sperm) or very early in development.


For a cancer to develop in Li-Fraumeni syndrome, a mutation involving the other copy of the TP53 gene must occur in the body's cells during a person's lifetime. Cells with two altered copies of this gene do not make functional TP53 protein, which allows tumors to develop. Almost everyone who inherits one TP53 gene mutation will eventually acquire a mutation in the second copy of the gene in some cells. The second mutation often occurs in cells within the breast, bone, or muscle tissue, typically leading to the tumors common in Li-Fraumeni syndrome.
Other Names for This Condition
- LFS
- Sarcoma family syndrome of Li and Fraumeni
- Sarcoma, breast, leukemia, and adrenal gland (SBLA) syndrome
- SBLA syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Batalini F, Peacock EG, Stobie L, Robertson A, Garber J, Weitzel JN, Tung NM. Li-Fraumeni syndrome: not a straightforward diagnosis anymore-the interpretation of pathogenic variants of low allele frequency and the differences between germline PVs, mosaicism, and clonal hematopoiesis. Breast Cancer Res. 2019 Sep 18;21(1):107. doi: 10.1186/s13058-019-1193-1. Citation on PubMed or Free article on PubMed Central
- Chompret A. The Li-Fraumeni syndrome. Biochimie. 2002 Jan;84(1):75-82. doi: 10.1016/s0300-9084(01)01361-x. Citation on PubMed
- Gargallo P, Yanez Y, Segura V, Juan A, Torres B, Balaguer J, Oltra S, Castel V, Canete A. Li-Fraumeni syndrome heterogeneity. Clin Transl Oncol. 2020 Jul;22(7):978-988. doi: 10.1007/s12094-019-02236-2. Epub 2019 Nov 5. Citation on PubMed
- Kratz CP, Achatz MI, Brugieres L, Frebourg T, Garber JE, Greer MC, Hansford JR, Janeway KA, Kohlmann WK, McGee R, Mullighan CG, Onel K, Pajtler KW, Pfister SM, Savage SA, Schiffman JD, Schneider KA, Strong LC, Evans DGR, Wasserman JD, Villani A, Malkin D. Cancer Screening Recommendations for Individuals with Li-Fraumeni Syndrome. Clin Cancer Res. 2017 Jun 1;23(11):e38-e45. doi: 10.1158/1078-0432.CCR-17-0408. Citation on PubMed
- Melean G, Sestini R, Ammannati F, Papi L. Genetic insights into familial tumors of the nervous system. Am J Med Genet C Semin Med Genet. 2004 Aug 15;129C(1):74-84. doi: 10.1002/ajmg.c.30022. Citation on PubMed
- Moule RN, Jhavar SG, Eeles RA. Genotype phenotype correlation in Li-Fraumeni syndrome kindreds and its implications for management. Fam Cancer. 2006;5(2):129-33. doi: 10.1007/s10689-005-4522-8. No abstract available. Citation on PubMed
- Olivier M, Goldgar DE, Sodha N, Ohgaki H, Kleihues P, Hainaut P, Eeles RA. Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2003 Oct 15;63(20):6643-50. Citation on PubMed
- Strong LC. General keynote: hereditary cancer: lessons from Li-Fraumeni syndrome. Gynecol Oncol. 2003 Jan;88(1 Pt 2):S4-7; discussion S11-3. doi: 10.1006/gyno.2002.6673. No abstract available. Citation on PubMed
- Varley JM. Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat. 2003 Mar;21(3):313-20. doi: 10.1002/humu.10185. Citation on PubMed
- Wong P, Verselis SJ, Garber JE, Schneider K, DiGianni L, Stockwell DH, Li FP, Syngal S. Prevalence of early onset colorectal cancer in 397 patients with classic Li-Fraumeni syndrome. Gastroenterology. 2006 Jan;130(1):73-9. doi: 10.1053/j.gastro.2005.10.014. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.