

Description
Hereditary cerebral amyloid angiopathy is a condition characterized by an abnormal buildup of protein clumps called amyloid deposits in the blood vessels in the brain, causing vascular disease (angiopathy). People with hereditary cerebral amyloid angiopathy often have progressive loss of intellectual function (dementia), stroke, and other neurological problems starting in mid-adulthood. Due to neurological decline, this condition is typically fatal in one's sixties, although there is variation depending on the severity of the signs and symptoms. Most affected individuals die within a decade after signs and symptoms first appear, although some people with the disease have survived longer.
There are many different types of hereditary cerebral amyloid angiopathy. The different types are distinguished by their genetic cause, which determines whether areas of the brain other than blood vessels are affected, and the signs and symptoms that occur. The various types of hereditary cerebral amyloid angiopathy are named after the regions where they were first diagnosed.
The Dutch type of hereditary cerebral amyloid angiopathy is the most common form. Stroke is frequently the first sign of the Dutch type and is fatal in about one third of people who have this condition. Survivors often develop dementia and have recurrent strokes. About half of individuals with the Dutch type who have one or more strokes will have recurrent seizures (epilepsy).
People with the Flemish and Italian types of hereditary cerebral amyloid angiopathy are prone to recurrent strokes and dementia. Individuals with the Piedmont type may have one or more strokes and typically experience impaired movements, numbness or tingling (paresthesias), confusion, or dementia.
The first sign of the Icelandic type of hereditary cerebral amyloid angiopathy is typically a stroke followed by dementia. Strokes associated with the Icelandic type usually occur earlier than the other types, with individuals typically experiencing their first stroke in their twenties or thirties.
Strokes are rare in people with the Arctic type of hereditary cerebral amyloid angiopathy, in which the first sign is usually memory loss that then progresses to severe dementia. Strokes are also uncommon in individuals with the Iowa type. This type is characterized by memory loss, problems with vocabulary and the production of speech, personality changes, and involuntary muscle twitches (myoclonus).
Two types of hereditary cerebral amyloid angiopathy, known as familial British dementia and familial Danish dementia, are characterized by dementia and movement problems. Strokes are uncommon in these types. People with the Danish type also have clouding of the lens of the eyes (cataracts ) and deafness.
) and deafness.
Frequency
The prevalence of hereditary cerebral amyloid angiopathy is unknown. The Dutch type is the most common, with over 200 affected individuals reported in the scientific literature.
Causes
Variants (also called mutations) in the APP gene are the most common cause of hereditary cerebral amyloid angiopathy. APP gene variants cause the Dutch, Italian, Arctic, Iowa, Flemish, and Piedmont types of this condition. Variants in the CST3 gene cause the Icelandic type. Familial British and Danish dementia are caused by variants in the ITM2B gene.
The APP gene provides instructions for making a protein called amyloid precursor protein. This protein is found in many tissues and organs, including the brain and spinal cord (central nervous system). The precise function of this protein is unknown, but researchers speculate that it may attach (bind) to other proteins on the surface of cells or help cells attach to one another. In the brain, the amyloid precursor protein plays a role in the development and maintenance of nerve cells (neurons).
The CST3 gene provides instructions for making a protein called cystatin C. This protein inhibits the activity of enzymes called cathepsins that cut apart other proteins in order to break them down. Cystatin C is found in biological fluids, such as blood. Its levels are especially high in the fluid that surrounds and protects the brain and spinal cord (the cerebrospinal fluid or CSF ).
).
The ITM2B gene provides instructions for producing a protein that is found in all tissues. The function of the ITM2B protein is unclear. It is thought to play a role in triggering the self-destruction of cells (apoptosis ) and keeping cells from growing and dividing too fast or in an uncontrolled way. Additionally, the ITM2B protein may be involved in processing the amyloid precursor protein.
) and keeping cells from growing and dividing too fast or in an uncontrolled way. Additionally, the ITM2B protein may be involved in processing the amyloid precursor protein.
Variants in the APP, CST3, or ITM2B gene lead to the production of proteins that are less stable than normal and that tend to cluster together (aggregate). These aggregated proteins form amyloid deposits that accumulate in certain areas of the brain and in its blood vessels . The amyloid deposits in the brain damage neurons, eventually causing cell death and impairing various parts of the brain. Brain cell loss in people with hereditary cerebral amyloid angiopathy can lead to seizures, movement abnormalities, and other neurological problems. In blood vessels
. The amyloid deposits in the brain damage neurons, eventually causing cell death and impairing various parts of the brain. Brain cell loss in people with hereditary cerebral amyloid angiopathy can lead to seizures, movement abnormalities, and other neurological problems. In blood vessels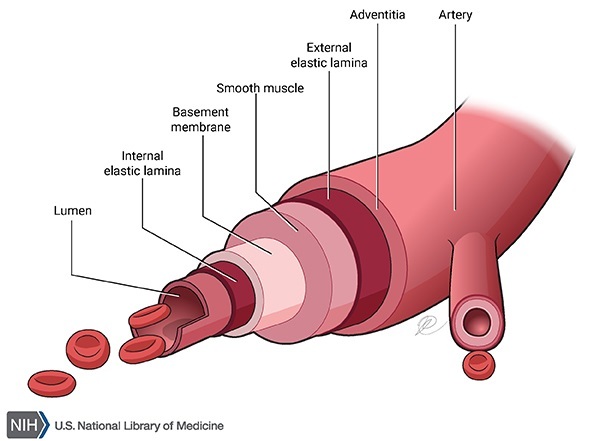 , amyloid deposits replace the muscle fibers and elastic fibers that give the blood vessels flexibility, causing them to become weak and prone to breakage. A break in a blood vessel in the brain causes bleeding in the brain (hemorrhagic stroke
, amyloid deposits replace the muscle fibers and elastic fibers that give the blood vessels flexibility, causing them to become weak and prone to breakage. A break in a blood vessel in the brain causes bleeding in the brain (hemorrhagic stroke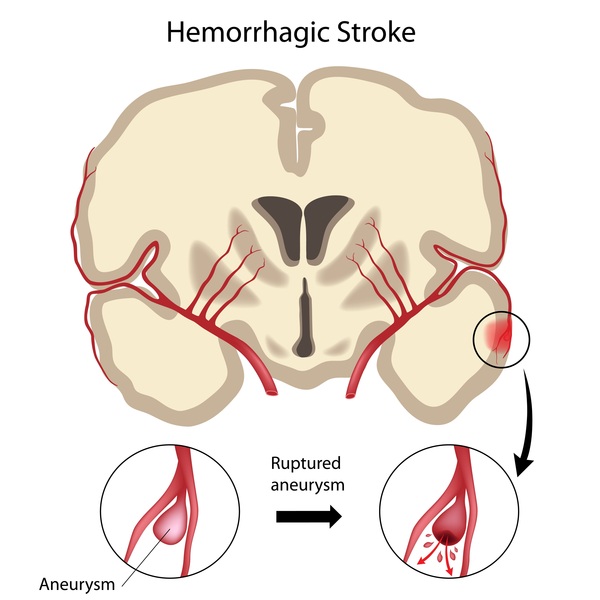 ), which can lead to brain damage and dementia or be life-threatening.
), which can lead to brain damage and dementia or be life-threatening.
Inheritance
Hereditary cerebral amyloid angiopathy caused by mutations in the APP, CST3, or ITM2B gene is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

There is also a non-hereditary form of cerebral amyloid angiopathy that occurs in people with no history of the disorder in their family. The cause of this form of the condition is unknown. These cases are described as sporadic and are not inherited.
Other Names for This Condition
- Autosomal dominant cerebrovascular amyloidosis
- CAA, familial
- Cerebral amyloid angiopathy, familial
- Cerebral amyloid angiopathy, genetic
- HCHWA-D
- HCHWA-I
- Hereditary cerebral hemorrhage with amyloidosis-Dutch type
- Hereditary cerebral hemorrhage with amyloidosis-Icelandic type
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Maat-Schieman M, Roos R, van Duinen S. Hereditary cerebral hemorrhage with amyloidosis-Dutch type. Neuropathology. 2005 Dec;25(4):288-97. doi: 10.1111/j.1440-1789.2005.00631.x. Citation on PubMed
- Maia LF, Mackenzie IR, Feldman HH. Clinical phenotypes of Cerebral Amyloid Angiopathy. J Neurol Sci. 2007 Jun 15;257(1-2):23-30. doi: 10.1016/j.jns.2007.01.054. Epub 2007 Mar 6. Citation on PubMed
- Murakami K, Irie K, Morimoto A, Ohigashi H, Shindo M, Nagao M, Shimizu T, Shirasawa T. Neurotoxicity and physicochemical properties of Abeta mutant peptides from cerebral amyloid angiopathy: implication for the pathogenesis of cerebral amyloid angiopathy and Alzheimer's disease. J Biol Chem. 2003 Nov 14;278(46):46179-87. doi: 10.1074/jbc.M301874200. Epub 2003 Aug 27. Citation on PubMed
- Palsdottir A, Snorradottir AO, Thorsteinsson L. Hereditary cystatin C amyloid angiopathy: genetic, clinical, and pathological aspects. Brain Pathol. 2006 Jan;16(1):55-9. doi: 10.1111/j.1750-3639.2006.tb00561.x. Citation on PubMed
- Pezzini A, Del Zotto E, Volonghi I, Giossi A, Costa P, Padovani A. Cerebral amyloid angiopathy: a common cause of cerebral hemorrhage. Curr Med Chem. 2009;16(20):2498-513. doi: 10.2174/092986709788682047. Citation on PubMed
- Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL. Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol. 2003 Sep;62(9):885-98. doi: 10.1093/jnen/62.9.885. Citation on PubMed
- Revesz T, Holton JL, Lashley T, Plant G, Frangione B, Rostagno A, Ghiso J. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 2009 Jul;118(1):115-30. doi: 10.1007/s00401-009-0501-8. Epub 2009 Feb 19. Citation on PubMed or Free article on PubMed Central
- Tomidokoro Y, Rostagno A, Neubert TA, Lu Y, Rebeck GW, Frangione B, Greenberg SM, Ghiso J. Iowa variant of familial Alzheimer's disease: accumulation of posttranslationally modified AbetaD23N in parenchymal and cerebrovascular amyloid deposits. Am J Pathol. 2010 Apr;176(4):1841-54. doi: 10.2353/ajpath.2010.090636. Epub 2010 Mar 12. Citation on PubMed or Free article on PubMed Central
- Zhang-Nunes SX, Maat-Schieman ML, van Duinen SG, Roos RA, Frosch MP, Greenberg SM. The cerebral beta-amyloid angiopathies: hereditary and sporadic. Brain Pathol. 2006 Jan;16(1):30-9. doi: 10.1111/j.1750-3639.2006.tb00559.x. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.