

Description
Dilated cardiomyopathy is a form of heart disease in which the heart (cardiac) muscle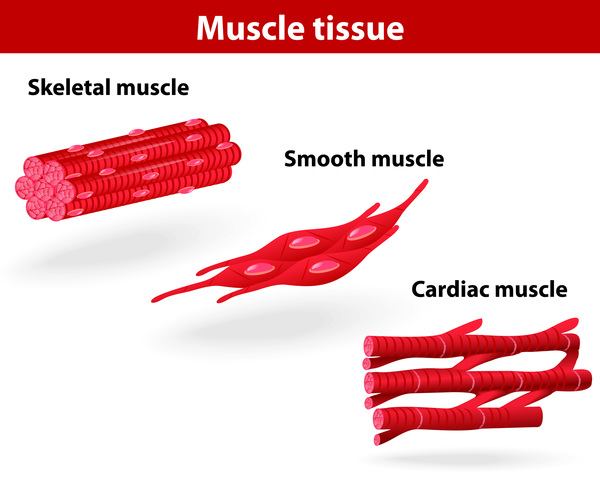 becomes thin and enlarged (dilated). The dilation, which typically starts in the lower left chamber of the heart (left ventricle), makes it harder for the heart to pump blood to the rest of the body.
becomes thin and enlarged (dilated). The dilation, which typically starts in the lower left chamber of the heart (left ventricle), makes it harder for the heart to pump blood to the rest of the body.
Dilated cardiomyopathy is called nonsyndromic dilated cardiomyopathy when it cannot be explained by other causes, such as a heart attack or damage to the valves of the heart, and is not associated with signs and symptoms that affect other parts of the body.
The signs and symptoms of nonsyndromic dilated cardiomyopathy vary among affected individuals, even among members of the same family. The signs and symptoms typically begin in mid-adulthood, but they can occur at any time from infancy to late adulthood. Affected individuals may have a sensation of fluttering or pounding in the chest (palpitations); shortness of breath, especially when lying down or during physical activity; fatigue; and swelling of the legs and feet. Affected individuals may also have episodes of dizziness or fainting (syncope).
Over time, people with nonsyndromic dilated cardiomyopathy may develop life-threatening complications, which can include abnormal heart rhythms (arrhythmias) and heart failure. Although uncommon, sudden death can occur in people with nonsyndromic dilated cardiomyopathy, even if they have no other symptoms of the condition.
Frequency
Dilated cardiomyopathy may affect as many as 1 in 250 people. In approximately 30 to 50 percent of these cases, there is a family history of the condition.
Causes
Variants (also called mutations) in more than 20 genes have been found to cause nonsyndromic dilated cardiomyopathy . These genes provide instructions for making proteins that are found in cardiac muscle cells called cardiomyocytes.
. These genes provide instructions for making proteins that are found in cardiac muscle cells called cardiomyocytes.
Many of these proteins play important roles in the tensing (contraction) of the cardiac muscle through their association with structures called sarcomeres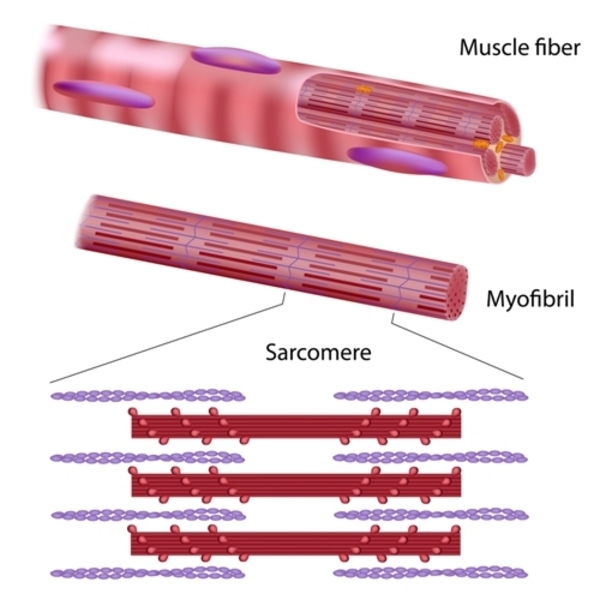 . Sarcomeres are made of proteins that generate the mechanical force needed for muscles to contract; they are the basic units of contraction. Other proteins that are associated with nonsyndromic dilated cardiomyopathy make up the structural framework (the cytoskeleton) of cardiomyocytes. The remaining proteins have various roles within cardiomyocytes to ensure that these cells function properly.
. Sarcomeres are made of proteins that generate the mechanical force needed for muscles to contract; they are the basic units of contraction. Other proteins that are associated with nonsyndromic dilated cardiomyopathy make up the structural framework (the cytoskeleton) of cardiomyocytes. The remaining proteins have various roles within cardiomyocytes to ensure that these cells function properly.
A genetic cause is found in approximately 25 to 35 percent of people with dilated cardiomyopathy. A genetic cause is more likely to be found in individuals who have affected family members.
Variants in one gene, TTN, account for approximately 15 to 20 percent of all cases of dilated cardiomyopathy. The TTN gene provides instructions for making a protein called titin, which is found in the sarcomeres of cardiomyocytes and other muscle cells. Titin provides structure, flexibility, and stability to sarcomeres. Titin also plays a role in chemical signaling and in the assembly of new sarcomeres. Many of the TTN gene variants in people with nonsyndromic dilated cardiomyopathy cause cells to produce an abnormally short version of the titin protein. The altered protein likely impairs sarcomere function and disrupts chemical signaling.
Dilated cardiomyopathy may also occur as part of a syndrome that affects other organs and tissues in the body. These forms of the condition are described as "syndromic" and are caused by variants in other genes.
Some cases of dilated cardiomyopathy are acquired during a person’s lifetime. Causes of acquired dilated cardiomyopathy can include damage to the valves that control the flow of blood through the heart and damage to the cardiac muscle that is caused by a heart attack, infections, or certain medications.
Inheritance
Nonsyndromic dilated cardiomyopathy has different inheritance patterns depending on the specific gene involved. When nonsyndromic dilated cardiomyopathy is present in multiple family members, it is often called familial dilated cardiomyopathy.
Nonsyndromic dilated cardiomyopathy is usually inherited in an autosomal dominant pattern , which means one copy of an altered gene in each cell is sufficient to cause the disorder. However, some people who have an altered gene never develop features of the condition. This is known as reduced penetrance.
, which means one copy of an altered gene in each cell is sufficient to cause the disorder. However, some people who have an altered gene never develop features of the condition. This is known as reduced penetrance.
Although many individuals with nonsyndromic dilated cardiomyopathy have an affected parent, some cases of this condition result from new (de novo) variants in a gene that occur during the formation of reproductive cells (eggs or sperm) in an affected individual's parent or during early embryonic development.
Nonsyndromic dilated cardiomyopathy can also be inherited in an autosomal recessive pattern , which means both copies of a gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of a gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Syndromic dilated cardiomyopathy follows the inheritance pattern of the associated syndrome.
Other Names for This Condition
- DCM
- Familial dilated cardiomyopathy
- Idiopathic dilated cardiomyopathy
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Dilated cardiomyopathy 1C

- Genetic Testing Registry: Dilated cardiomyopathy 1CC
- Genetic Testing Registry: Dilated cardiomyopathy 1DD
- Genetic Testing Registry: Dilated cardiomyopathy 1E
- Genetic Testing Registry: Dilated cardiomyopathy 1FF
- Genetic Testing Registry: Dilated cardiomyopathy 1G
- Genetic Testing Registry: Dilated cardiomyopathy 1HH
- Genetic Testing Registry: Dilated cardiomyopathy 1I
- Genetic Testing Registry: Dilated cardiomyopathy 1JJ
- Genetic Testing Registry: Dilated cardiomyopathy 1R
- Genetic Testing Registry: Dilated cardiomyopathy 1S
- Genetic Testing Registry: Dilated cardiomyopathy 1V
- Genetic Testing Registry: Dilated cardiomyopathy 1W
- Genetic Testing Registry: Dilated cardiomyopathy 1Y
- Genetic Testing Registry: Dilated cardiomyopathy 1Z
- Genetic Testing Registry: Dilated cardiomyopathy 2A
- Genetic Testing Registry: Dilated cardiomyopathy 1A
- Genetic Testing Registry: Dilated cardiomyopathy 1B
- Genetic Testing Registry: Dilated cardiomyopathy 1D
- Genetic Testing Registry: Dilated cardiomyopathy 1K
- Genetic Testing Registry: Dilated cardiomyopathy 1U
- Genetic Testing Registry: Familial isolated dilated cardiomyopathy
- Genetic Testing Registry: Primary dilated cardiomyopathy
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
- CARDIOMYOPATHY, DILATED, 1A; CMD1A
- CARDIOMYOPATHY, DILATED, 1C, WITH OR WITHOUT LEFT VENTRICULAR NONCOMPACTION; CMD1C
- CARDIOMYOPATHY, DILATED, 1D; CMD1D
- CARDIOMYOPATHY, DILATED, 1B; CMD1B
- CARDIOMYOPATHY, DILATED, 3B; CMD3B
- CARDIOMYOPATHY, DILATED, 1E; CMD1E
- CARDIOMYOPATHY, DILATED, 1I; CMD1I
- CARDIOMYOPATHY, DILATED, 1J; CMD1J
- CARDIOMYOPATHY, DILATED, 1G; CMD1G
- CARDIOMYOPATHY, DILATED, 1H; CMD1H
- CARDIOMYOPATHY, DILATED, 1O; CMD1O
- CARDIOMYOPATHY, DILATED, 1L; CMD1L
- CARDIOMYOPATHY, DILATED, 1K; CMD1K
- CARDIOMYOPATHY, DILATED, 1M; CMD1M
- CARDIOMYOPATHY, FAMILIAL HYPERTROPHIC, 25; CMH25
- CARDIOMYOPATHY, DILATED, 1P; CMD1P
- CARDIOMYOPATHY, DILATED, 1Q; CMD1Q
- CARDIOMYOPATHY, DILATED, 1W; CMD1W
- CARDIOMYOPATHY, DILATED, 1Y; CMD1Y
- CARDIOMYOPATHY, DILATED, 1Z; CMD1Z
- CARDIOMYOPATHY, DILATED, 2A; CMD2A
- CARDIOMYOPATHY, DILATED, 1AA, WITH OR WITHOUT LEFT VENTRICULAR NONCOMPACTION; CMD1AA
- LEFT VENTRICULAR NONCOMPACTION 10; LVNC10
- CARDIOMYOPATHY, DILATED, 1DD; CMD1DD
- CARDIOMYOPATHY, DILATED, 1KK; CMD1KK
- CARDIOMYOPATHY, DILATED, 1U; CMD1U
- CARDIOMYOPATHY, DILATED, 1V; CMD1V
- CARDIOMYOPATHY, DILATED, 1EE; CMD1EE
- CARDIOMYOPATHY, DILATED, 1R; CMD1R
- CARDIOMYOPATHY, DILATED, 1S; CMD1S
- CARDIOMYOPATHY, DILATED, 1JJ; CMD1JJ
- CARDIOMYOPATHY, DILATED, 1HH; CMD1HH
- CARDIOMYOPATHY, DILATED, 2B; CMD2B
- CARDIOMYOPATHY, DILATED, 1II; CMD1II
- CARDIOMYOPATHY, DILATED, 1BB; CMD1BB
Scientific Articles on PubMed
References
- Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012 Feb 16;366(7):619-28. doi: 10.1056/NEJMoa1110186. Citation on PubMed or Free article on PubMed Central
- Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, Morales A, Taylor MRG, Vatta M, Ware SM; ACMG Professional Practice and Guidelines Committee. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2018 Sep;20(9):899-909. doi: 10.1038/s41436-018-0039-z. Epub 2018 Jun 14. Citation on PubMed
- Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013 Sep;10(9):531-47. doi: 10.1038/nrcardio.2013.105. Epub 2013 Jul 30. Citation on PubMed
- Hershberger RE, Jordan E. Dilated Cardiomyopathy Overview. 2007 Jul 27 [updated 2024 Dec 12]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1309/ Citation on PubMed
- Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med. 2010 Nov;12(11):655-67. doi: 10.1097/GIM.0b013e3181f2481f. Citation on PubMed or Free article on PubMed Central
- Jordan E, Hershberger RE. Considering complexity in the genetic evaluation of dilated cardiomyopathy. Heart. 2021 Jan;107(2):106-112. doi: 10.1136/heartjnl-2020-316658. Epub 2020 Oct 27. Citation on PubMed
- Jordan E, Peterson L, Ai T, Asatryan B, Bronicki L, Brown E, Celeghin R, Edwards M, Fan J, Ingles J, James CA, Jarinova O, Johnson R, Judge DP, Lahrouchi N, Lekanne Deprez RH, Lumbers RT, Mazzarotto F, Medeiros Domingo A, Miller RL, Morales A, Murray B, Peters S, Pilichou K, Protonotarios A, Semsarian C, Shah P, Syrris P, Thaxton C, van Tintelen JP, Walsh R, Wang J, Ware J, Hershberger RE. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation. 2021 Jul 6;144(1):7-19. doi: 10.1161/CIRCULATIONAHA.120.053033. Epub 2021 May 5. Citation on PubMed
- McGurk KA, Zhang X, Theotokis P, Thomson K, Harper A, Buchan RJ, Mazaika E, Ormondroyd E, Wright WT, Macaya D, Pua CJ, Funke B, MacArthur DG, Prasad SK, Cook SA, Allouba M, Aguib Y, Yacoub MH, O'Regan DP, Barton PJR, Watkins H, Bottolo L, Ware JS. The penetrance of rare variants in cardiomyopathy-associated genes: A cross-sectional approach to estimating penetrance for secondary findings. Am J Hum Genet. 2023 Sep 7;110(9):1482-1495. doi: 10.1016/j.ajhg.2023.08.003. Epub 2023 Aug 30. Citation on PubMed
- McNally EM, Mestroni L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ Res. 2017 Sep 15;121(7):731-748. doi: 10.1161/CIRCRESAHA.116.309396. Citation on PubMed
- Musunuru K, Hershberger RE, Day SM, Klinedinst NJ, Landstrom AP, Parikh VN, Prakash S, Semsarian C, Sturm AC; American Heart Association Council on Genomic and Precision Medicine; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular and Stroke Nursing; and Council on Clinical Cardiology. Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement From the American Heart Association. Circ Genom Precis Med. 2020 Aug;13(4):e000067. doi: 10.1161/HCG.0000000000000067. Epub 2020 Jul 23. Citation on PubMed
- Schultheiss HP, Fairweather D, Caforio ALP, Escher F, Hershberger RE, Lipshultz SE, Liu PP, Matsumori A, Mazzanti A, McMurray J, Priori SG. Dilated cardiomyopathy. Nat Rev Dis Primers. 2019 May 9;5(1):32. doi: 10.1038/s41572-019-0084-1. Citation on PubMed
- Tayal U, Ware JS, Lakdawala NK, Heymans S, Prasad SK. Understanding the genetics of adult-onset dilated cardiomyopathy: what a clinician needs to know. Eur Heart J. 2021 Jun 21;42(24):2384-2396. doi: 10.1093/eurheartj/ehab286. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.