

Description
Cowden syndrome is a genetic disorder characterized by multiple noncancerous, tumor-like growths called hamartomas and an increased risk of developing certain cancers.
Almost everyone with Cowden syndrome develops hamartomas. These growths are most commonly found on the skin and mucous membranes (such as the lining of the mouth and nose), but they can also occur in the intestine and other parts of the body. The growth of hamartomas on the skin and mucous membranes typically becomes apparent by a person's late twenties.
Cowden syndrome is associated with an increased risk of developing several types of cancer, particularly cancers of the breast, a gland in the lower neck called the thyroid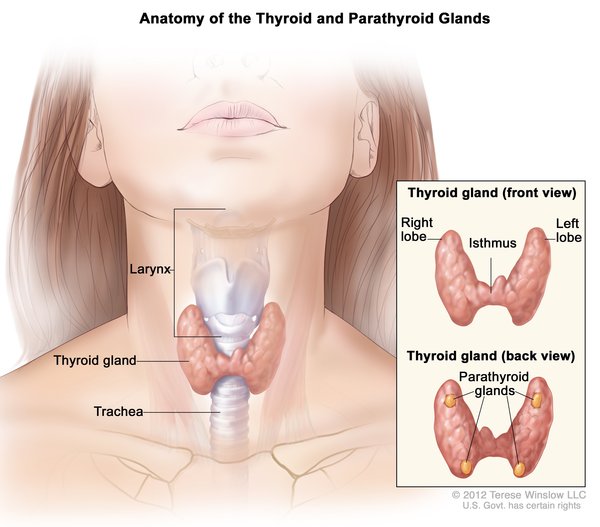 , and the lining of the uterus (the endometrium
, and the lining of the uterus (the endometrium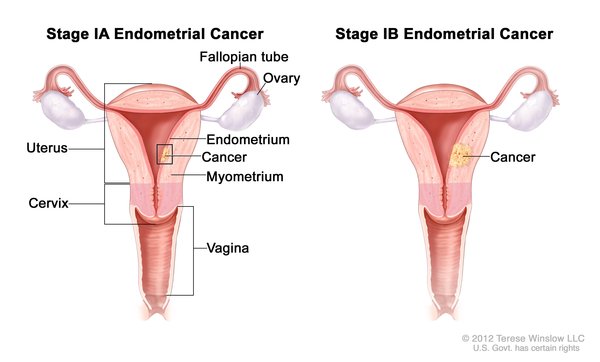 ). Other cancers that have been identified in people with Cowden syndrome include kidney cancer, colorectal cancer
). Other cancers that have been identified in people with Cowden syndrome include kidney cancer, colorectal cancer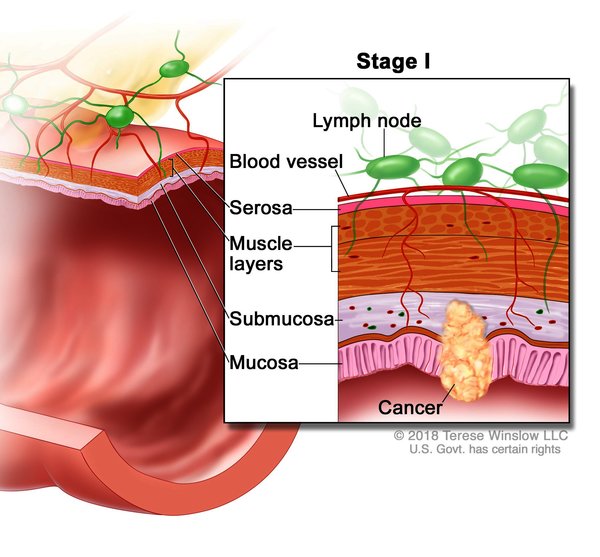 , and an agressive form of skin cancer called melanoma. Compared with the general population, people with Cowden syndrome develop these cancers at younger ages, often beginning in their thirties or forties. People with Cowden syndrome are also more likely to develop more than one cancer during their lifetimes compared to the general population. Other diseases of the breast, thyroid, and endometrium are also common in Cowden syndrome. Additional signs and symptoms can include an enlarged head (macrocephaly
, and an agressive form of skin cancer called melanoma. Compared with the general population, people with Cowden syndrome develop these cancers at younger ages, often beginning in their thirties or forties. People with Cowden syndrome are also more likely to develop more than one cancer during their lifetimes compared to the general population. Other diseases of the breast, thyroid, and endometrium are also common in Cowden syndrome. Additional signs and symptoms can include an enlarged head (macrocephaly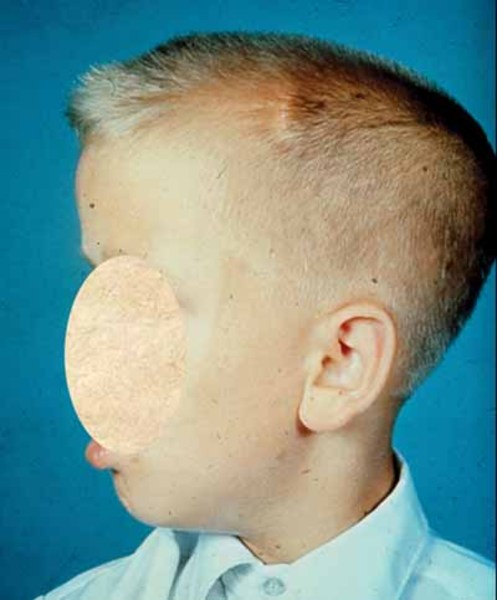 ) and a rare, noncancerous brain tumor called Lhermitte-Duclos disease. A small percentage of affected individuals have delayed development, intellectual disability, or autism spectrum disorder, which can affect communication and social interaction.
) and a rare, noncancerous brain tumor called Lhermitte-Duclos disease. A small percentage of affected individuals have delayed development, intellectual disability, or autism spectrum disorder, which can affect communication and social interaction.
Some people do not meet the strict criteria for a clinical diagnosis of Cowden syndrome, but they have some of the characteristic features of the condition, particularly the cancers. These individuals are often described as having Cowden-like syndrome. Both Cowden syndrome and Cowden-like syndrome are caused by mutations in the same genes.
The features of Cowden syndrome overlap with those of another disorder called Bannayan-Riley-Ruvalcaba syndrome. People with Bannayan-Riley-Ruvalcaba syndrome also develop hamartomas and other noncancerous tumors. Some people with Cowden syndrome have relatives diagnosed with Bannayan-Riley-Ruvalcaba syndrome, and other affected individuals have the characteristic features of both conditions. Based on these similarities, researchers have proposed that Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome represent a spectrum of overlapping features known as PTEN hamartoma tumor syndrome (named for the genetic cause of the conditions) instead of two distinct conditions.
Frequency
Although the exact prevalence of Cowden syndrome is unknown, researchers estimate that it affects about 1 in 200,000 people.
Causes
Changes in the PTEN, KLLN, or WWP1 gene are most commonly identified in people with Cowden syndrome or Cowden-like syndrome.
About 25 percent of Cowden syndrome and a small percentage of cases of Cowden-like syndrome result from mutations in the PTEN gene. The protein produced from the PTEN gene is a tumor suppressor, which means that it normally prevents cells from growing and dividing (proliferating) too rapidly or in an uncontrolled way
(proliferating) too rapidly or in an uncontrolled way . Mutations in the PTEN gene prevent the PTEN protein from regulating cell proliferation effectively, leading to uncontrolled cell division and the formation of hamartomas and cancerous tumors. The PTEN gene likely has other important functions within cells; however, research is needed to determine what role mutations in this gene play in causing the other features of Cowden syndrome, such as macrocephaly and intellectual disability.
. Mutations in the PTEN gene prevent the PTEN protein from regulating cell proliferation effectively, leading to uncontrolled cell division and the formation of hamartomas and cancerous tumors. The PTEN gene likely has other important functions within cells; however, research is needed to determine what role mutations in this gene play in causing the other features of Cowden syndrome, such as macrocephaly and intellectual disability.
Rarely, Cowden syndrome and Cowden-like syndrome result from a change involving the KLLN gene. This gene provides instructions for making a protein called killin. Like the protein produced from the PTEN gene, killin probably acts as a tumor suppressor. The genetic change that causes Cowden syndrome and Cowden-like syndrome leads to reduced production of the killin protein. A reduced amount of killin may allow abnormal cells to survive and proliferate inappropriately, which can lead to the formation of tumors.
A small percentage of Cowden syndrome and Cowden-like syndrome are associated with variants in the WWP1 gene. The WWP1 gene provides instructions for making a protein that is involved in the process that targets other proteins to be broken down (degraded) within cells. During this process, the WWP1 protein attaches (binds) to the PTEN protein, which impairs PTEN's function. WWP1 gene variants are described as "gain-of-function" because they appear to enhance the activity of the WWP1 protein. Studies suggest that the altered protein binds to the PTEN protein more readily than normal. Excessive binding impairs PTEN's tumor suppressor activity, allowing cells to proliferate unchecked and, leading to the formation of tumors.
Mutations in a few other genes are each responsible for a very small percentage of cases of Cowden syndrome and Cowden-like syndrome. In the remaining cases, the genetic cause is unknown.
Inheritance
Cowden syndrome and Cowden-like syndrome are inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the condition and increase the risk of developing cancer. In some cases, an affected person inherits the mutation from one affected parent. Other cases may result from new mutations in the gene. These cases occur in people with no history of the disorder in their family.


Other Names for This Condition
- CD
- Cowden disease
- Cowden's disease
- Cowden's syndrome
- CS
- MHAM
- Multiple hamartoma syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bennett KL, Mester J, Eng C. Germline epigenetic regulation of KILLIN in Cowden and Cowden-like syndrome. JAMA. 2010 Dec 22;304(24):2724-31. doi: 10.1001/jama.2010.1877. Citation on PubMed or Free article on PubMed Central
- Blumenthal GM, Dennis PA. PTEN hamartoma tumor syndromes. Eur J Hum Genet. 2008 Nov;16(11):1289-300. doi: 10.1038/ejhg.2008.162. Epub 2008 Sep 10. Citation on PubMed
- Hobert JA, Eng C. PTEN hamartoma tumor syndrome: an overview. Genet Med. 2009 Oct;11(10):687-94. doi: 10.1097/GIM.0b013e3181ac9aea. Citation on PubMed
- Lee YR, Yehia L, Kishikawa T, Ni Y, Leach B, Zhang J, Panch N, Liu J, Wei W, Eng C, Pandolfi PP. WWP1 Gain-of-Function Inactivation of PTEN in Cancer Predisposition. N Engl J Med. 2020 May 28;382(22):2103-2116. doi: 10.1056/NEJMoa1914919. Citation on PubMed
- Ni Y, He X, Chen J, Moline J, Mester J, Orloff MS, Ringel MD, Eng C. Germline SDHx variants modify breast and thyroid cancer risks in Cowden and Cowden-like syndrome via FAD/NAD-dependant destabilization of p53. Hum Mol Genet. 2012 Jan 15;21(2):300-10. doi: 10.1093/hmg/ddr459. Epub 2011 Oct 6. Citation on PubMed or Free article on PubMed Central
- Ni Y, Zbuk KM, Sadler T, Patocs A, Lobo G, Edelman E, Platzer P, Orloff MS, Waite KA, Eng C. Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am J Hum Genet. 2008 Aug;83(2):261-8. doi: 10.1016/j.ajhg.2008.07.011. Citation on PubMed or Free article on PubMed Central
- Tan MH, Mester J, Peterson C, Yang Y, Chen JL, Rybicki LA, Milas K, Pederson H, Remzi B, Orloff MS, Eng C. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet. 2011 Jan 7;88(1):42-56. doi: 10.1016/j.ajhg.2010.11.013. Epub 2010 Dec 30. Citation on PubMed or Free article on PubMed Central
- Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012 Jan 15;18(2):400-7. doi: 10.1158/1078-0432.CCR-11-2283. Citation on PubMed or Free article on PubMed Central
- Yehia L, Eng C. 65 YEARS OF THE DOUBLE HELIX: One gene, many endocrine and metabolic syndromes: PTEN-opathies and precision medicine. Endocr Relat Cancer. 2018 Aug;25(8):T121-T140. doi: 10.1530/ERC-18-0162. Epub 2018 May 23. Citation on PubMed
- Yehia L, Eng C. PTEN Hamartoma Tumor Syndrome. 2001 Nov 29 [updated 2025 Aug 7]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1488/ Citation on PubMed
- Yehia L, Eng C. PTEN hamartoma tumour syndrome: what happens when there is no PTEN germline mutation? Hum Mol Genet. 2020 Oct 20;29(R2):R150-R157. doi: 10.1093/hmg/ddaa127. Citation on PubMed
- Yehia L, Keel E, Eng C. The Clinical Spectrum of PTEN Mutations. Annu Rev Med. 2020 Jan 27;71:103-116. doi: 10.1146/annurev-med-052218-125823. Epub 2019 Aug 21. Citation on PubMed
- Yehia L, Ngeow J, Eng C. PTEN-opathies: from biological insights to evidence-based precision medicine. J Clin Invest. 2019 Feb 1;129(2):452-464. doi: 10.1172/JCI121277. Epub 2019 Jan 7. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.