

Description
Constitutional mismatch repair deficiency (CMMRD) syndrome is a rare disorder that greatly increases the risk of developing different types of cancer throughout a person's lifetime. Affected individuals often develop their first cancer in childhood. The cancers that most commonly occur in people with CMMRD are cancers of the colon and rectum (collectively referred to as colorectal cancer), blood (leukemia or lymphoma), and brain.
and rectum (collectively referred to as colorectal cancer), blood (leukemia or lymphoma), and brain.
Approximately 50 percent of people with CMMRD will develop cancer by age 10, and 90 percent will develop cancer by age 18. Brain cancers, leukemias, and lymphomas tend to occur at a younger age than colorectal cancer in affected individuals. Nearly all people with CMMRD will develop a second cancer if they survive the first cancer.
People with CMMRD often develop multiple abnormal growths (polyps) on the lining of the colon. If these polyps are not removed, they may become cancerous over time.
Brain cancers in people with CMMRD are often high-grade gliomas or glioblastomas, which are tumors that are made up of cells called glial cells.
The most common blood cancers in people with CMMRD are non-Hodgkin lymphomas, such as lymphoblastic lymphoma. These cancers primarily affect white blood cells known as T cells.
such as lymphoblastic lymphoma. These cancers primarily affect white blood cells known as T cells.
Other cancers that can occur in people who have CMMRD include cancers of the small intestine , urinary tract
, urinary tract , or lining of the uterus. Cancers of the connective tissue and bone (sarcomas) may also develop.
, or lining of the uterus. Cancers of the connective tissue and bone (sarcomas) may also develop.
Some affected individuals have patches of skin that are unusually light in color (hypopigmented). Many people with CMMRD develop features similar to those seen in people with a condition called neurofibromatosis type 1. These features include changes in skin coloring (pigmentation), which are characterized by multiple flat patches on the skin that are darker than the surrounding area (café-au-lait spots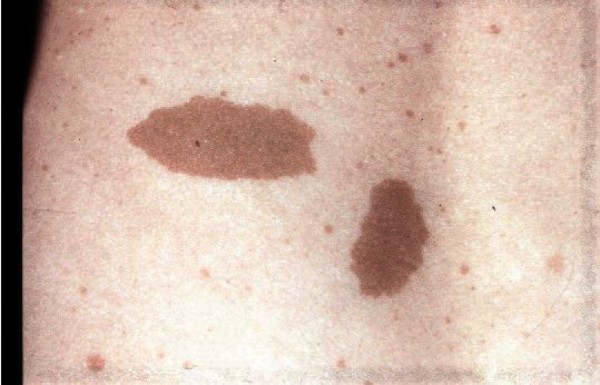 ). Because of these shared features, CMMRD is sometimes initially misdiagnosed as neurofibromatosis type 1.
). Because of these shared features, CMMRD is sometimes initially misdiagnosed as neurofibromatosis type 1.
Many people with CMMRD also develop a noncancerous (benign) feature called developmental venous anomaly, which is a rearrangement of the small veins in the brain. This feature can only be seen with medical imaging.
Due to the young age at which people with CMMRD develop cancer, the lifespan of affected individuals is typically shortened, with many people surviving only into adolescence or early adulthood.
Frequency
CMMRD is a rare disorder. Only a few hundred affected individuals have been reported in the medical literature. The incidence of CMMRD is estimated to be about 1 in 1,000,000 newborns. The incidence is likely higher among people whose parents are related to one another.
Causes
Genetic changes that cause disease or increase the risk of disease are sometimes called mutations or pathogenic variants. Pathogenic variants in the PMS2 gene cause more than 50 percent of cases of CMMRD, while pathogenic variants in the MSH6 gene cause approximately 20 percent of cases. Pathogenic variants in the MLH1 and MSH2 genes are responsible for the remaining cases of CMMRD. These four genes are involved in repairing errors that occur when DNA is copied in preparation for cell division, a process called DNA replication . Because these genes work together to fix DNA errors, they are known as mismatch repair (MMR) genes.
. Because these genes work together to fix DNA errors, they are known as mismatch repair (MMR) genes.
The pathogenic variants that cause CMMRD reduce the amount of functional MMR protein. A shortage of any of the MMR proteins impairs the cell's ability to fix the errors that naturally occur during DNA replication. These errors can then accumulate and disrupt other genes that are involved in important cellular processes such as cell growth and cell division (proliferation). Uncontrolled cell growth leads to cancer.
Particular pathogenic variants in MMR genes cause a form of CMMRD called attenuated CMMRD. One such pathogenic variant in the PMS2 gene is found in the Nunavik population of Quebec, Canada. People with attenuated CMMRD typically develop cancer later in life, often in adulthood.
Inheritance
CMMRD is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a pathogenic variant to cause the disorder.
, which means both copies of the gene in each cell must have a pathogenic variant to cause the disorder.
The parents of an individual with CMMRD typically have a pathogenic variant in one copy of a gene associated with CMMRD in each of their cells. These parents have a cancer predisposition syndrome called Lynch syndrome. Lynch syndrome increases the risk of many types of cancer, particularly colorectal cancer, but also cancers of the stomach, small intestine, gallbladder ducts, upper urinary tract, ovaries, endometrium, brain, and skin. Unlike CMMRD, individuals with Lynch syndrome develop these cancers in adulthood. People with CMMRD may not have a history of cancer in their family, as some people with a pathogenic variant in only one copy of an MMR gene do not develop cancer.
Other Names for This Condition
- CMMRD
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Aronson M, Colas C, Shuen A, Hampel H, Foulkes WD, Baris Feldman H, Goldberg Y, Muleris M, Wolfe Schneider K, McGee RB, Jasperson K, Rangaswami A, Brugieres L, Tabori U. Diagnostic criteria for constitutional mismatch repair deficiency (CMMRD): recommendations from the international consensus working group. J Med Genet. 2022 Apr;59(4):318-327. doi: 10.1136/jmedgenet-2020-107627. Epub 2021 Feb 23. Citation on PubMed
- Colas C, Guerrini-Rousseau L, Suerink M, Gallon R, Kratz CP, Ayuso E; ERN GENTURIS CMMRD Guideline Group; Brugieres L, Wimmer K. ERN GENTURIS guidelines on constitutional mismatch repair deficiency diagnosis, genetic counselling, surveillance, quality of life, and clinical management. Eur J Hum Genet. 2024 Dec;32(12):1526-1541. doi: 10.1038/s41431-024-01708-6. Epub 2024 Oct 17. Citation on PubMed
- Durno C, Boland CR, Cohen S, Dominitz JA, Giardiello FM, Johnson DA, Kaltenbach T, Levin TR, Lieberman D, Robertson DJ, Rex DK. Recommendations on Surveillance and Management of Biallelic Mismatch Repair Deficiency (BMMRD) Syndrome: A Consensus Statement by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2017 May;152(6):1605-1614. doi: 10.1053/j.gastro.2017.02.011. Epub 2017 Mar 28. Citation on PubMed
- Durno C, Ercan AB, Bianchi V, Edwards M, Aronson M, Galati M, Atenafu EG, Abebe-Campino G, Al-Battashi A, Alharbi M, Azad VF, Baris HN, Basel D, Bedgood R, Bendel A, Ben-Shachar S, Blumenthal DT, Blundell M, Bornhorst M, Bronsema A, Cairney E, Rhode S, Caspi S, Chamdin A, Chiaravalli S, Constantini S, Crooks B, Das A, Dvir R, Farah R, Foulkes WD, Frenkel Z, Gallinger B, Gardner S, Gass D, Ghalibafian M, Gilpin C, Goldberg Y, Goudie C, Hamid SA, Hampel H, Hansford JR, Harlos C, Hijiya N, Hsu S, Kamihara J, Kebudi R, Knipstein J, Koschmann C, Kratz C, Larouche V, Lassaletta A, Lindhorst S, Ling SC, Link MP, Loret De Mola R, Luiten R, Lurye M, Maciaszek JL, MagimairajanIssai V, Maher OM, Massimino M, McGee RB, Mushtaq N, Mason G, Newmark M, Nicholas G, Nichols KE, Nicolaides T, Opocher E, Osborn M, Oshrine B, Pearlman R, Pettee D, Rapp J, Rashid M, Reddy A, Reichman L, Remke M, Robbins G, Roy S, Sabel M, Samuel D, Scheers I, Schneider KW, Sen S, Stearns D, Sumerauer D, Swallow C, Taylor L, Thomas G, Toledano H, Tomboc P, Van Damme A, Winer I, Yalon M, Yen LY, Zapotocky M, Zelcer S, Ziegler DS, Zimmermann S, Hawkins C, Malkin D, Bouffet E, Villani A, Tabori U. Survival Benefit for Individuals With Constitutional Mismatch Repair Deficiency Undergoing Surveillance. J Clin Oncol. 2021 Sep 1;39(25):2779-2790. doi: 10.1200/JCO.20.02636. Epub 2021 May 4. Citation on PubMed
- Ercan AB, Aronson M, Fernandez NR, Chang Y, Levine A, Liu ZA, Negm L, Edwards M, Bianchi V, Stengs L, Chung J, Al-Battashi A, Reschke A, Lion A, Ahmad A, Lassaletta A, Reddy AT, Al-Darraji AF, Shah AC, Van Damme A, Bendel A, Rashid A, Margol AS, Kelly BL, Pencheva B, Heald B, Lemieux-Anglin B, Crooks B, Koschmann C, Gilpin C, Porter CC, Gass D, Samuel D, Ziegler DS, Blumenthal DT, Kuo DJ, Hamideh D, Basel D, Khuong-Quang DA, Stearns D, Opocher E, Carceller F, Baris Feldman H, Toledano H, Winer I, Scheers I, Fedorakova I, Su JM, Vengoechea J, Sterba J, Knipstein J, Hansford JR, Gonzales-Santos JR, Bhatia K, Bielamowicz KJ, Minhas K, Nichols KE, Cole KA, Penney L, Hjort MA, Sabel M, Gil-da-Costa MJ, Murray MJ, Miller M, Blundell ML, Massimino M, Al-Hussaini M, Al-Jadiry MF, Comito MA, Osborn M, Link MP, Zapotocky M, Ghalibafian M, Shaheen N, Mushtaq N, Waespe N, Hijiya N, Fuentes-Bolanos N, Ahmad O, Chamdine O, Roy P, Pichurin PN, Nyman P, Pearlman R, Auer RC, Sukumaran RK, Kebudi R, Dvir R, Raphael R, Elhasid R, McGee RB, Chami R, Noss R, Tanaka R, Raskin S, Sen S, Lindhorst S, Perreault S, Caspi S, Riaz S, Constantini S, Albert S, Chaleff S, Bielack S, Chiaravalli S, Cramer SL, Roy S, Cahn S, Penna S, Hamid SA, Ghafoor T, Imam U, Larouche V, Magimairajan Issai V, Foulkes WD, Lee YY, Nathan PC, Maruvka YE, Greer MC, Durno C, Shlien A, Ertl-Wagner B, Villani A, Malkin D, Hawkins C, Bouffet E, Das A, Tabori U. Clinical and biological landscape of constitutional mismatch-repair deficiency syndrome: an International Replication Repair Deficiency Consortium cohort study. Lancet Oncol. 2024 May;25(5):668-682. doi: 10.1016/S1470-2045(24)00026-3. Epub 2024 Mar 26. Citation on PubMed
- Gallon R, Brekelmans C, Martin M, Bours V, Schamschula E, Amberger A, Muleris M, Colas C, Dekervel J, De Hertogh G, Coupier J, Colleye O, Sepulchre E, Burn J, Brems H, Legius E, Wimmer K. Constitutional mismatch repair deficiency mimicking Lynch syndrome is associated with hypomorphic mismatch repair gene variants. NPJ Precis Oncol. 2024 May 24;8(1):119. doi: 10.1038/s41698-024-00603-z. Citation on PubMed
- Guerrini-Rousseau L, Gallon R, Pineda M, Brugieres L, Baert-Desurmont S, Corsini C, Dangouloff-Ros V, Gorris MAJ, Haberler C, Hoarau P, Jongmans MC, Kloor M, Loeffen J, Rigaud C, Robbe J, Vibert R, Weijers D, Wimmer K, Colas C; Care For CMMRD consortium. Report of the sixth meeting of the European Consortium 'Care for CMMRD' (C4CMMRD), Paris, France, November 16th 2022. Fam Cancer. 2024 Nov;23(4):447-457. doi: 10.1007/s10689-024-00403-1. Epub 2024 Jul 20. Citation on PubMed
- Guerrini-Rousseau L, Pasmant E, Muleris M, Abbou S, Adam-De-Beaumais T, Brugieres L, Cabaret O, Colas C, Cotteret S, Decq P, Dufour C, Guillerm E, Rouleau E, Varlet P, Zili S, Vidaud D, Grill J. Neurofibromatosis type 1 mosaicism in patients with constitutional mismatch repair deficiency. J Med Genet. 2024 Jan 19;61(2):158-162. doi: 10.1136/jmg-2023-109235. Citation on PubMed
- Lavoine N, Colas C, Muleris M, Bodo S, Duval A, Entz-Werle N, Coulet F, Cabaret O, Andreiuolo F, Charpy C, Sebille G, Wang Q, Lejeune S, Buisine MP, Leroux D, Couillault G, Leverger G, Fricker JP, Guimbaud R, Mathieu-Dramard M, Jedraszak G, Cohen-Hagenauer O, Guerrini-Rousseau L, Bourdeaut F, Grill J, Caron O, Baert-Dusermont S, Tinat J, Bougeard G, Frebourg T, Brugieres L. Constitutional mismatch repair deficiency syndrome: clinical description in a French cohort. J Med Genet. 2015 Nov;52(11):770-8. doi: 10.1136/jmedgenet-2015-103299. Epub 2015 Aug 28. Citation on PubMed
- Li L, Hamel N, Baker K, McGuffin MJ, Couillard M, Gologan A, Marcus VA, Chodirker B, Chudley A, Stefanovici C, Durandy A, Hegele RA, Feng BJ, Goldgar DE, Zhu J, De Rosa M, Gruber SB, Wimmer K, Young B, Chong G, Tischkowitz MD, Foulkes WD. A homozygous PMS2 founder mutation with an attenuated constitutional mismatch repair deficiency phenotype. J Med Genet. 2015 May;52(5):348-52. doi: 10.1136/jmedgenet-2014-102934. Epub 2015 Feb 17. Citation on PubMed
- Raveneau M, Guerrini-Rousseau L, Levy R, Roux CJ, Bolle S, Doz F, Bourdeaut F, Colas C, Blauwblomme T, Beccaria K, Tauziede-Espariat A, Varlet P, Dufour C, Grill J, Boddaert N, Dangouloff-Ros V. Specific brain MRI features of constitutional mismatch repair deficiency syndrome in children with high-grade gliomas. Eur Radiol. 2024 Dec;34(12):7765-7775. doi: 10.1007/s00330-024-10885-3. Epub 2024 Jul 9. Citation on PubMed
- Sijmons RH, Hofstra RMW. Review: Clinical aspects of hereditary DNA Mismatch repair gene mutations. DNA Repair (Amst). 2016 Feb;38:155-162. doi: 10.1016/j.dnarep.2015.11.018. Epub 2015 Dec 11. Citation on PubMed
- Wimmer K, Kratz CP, Vasen HF, Caron O, Colas C, Entz-Werle N, Gerdes AM, Goldberg Y, Ilencikova D, Muleris M, Duval A, Lavoine N, Ruiz-Ponte C, Slavc I, Burkhardt B, Brugieres L; EU-Consortium Care for CMMRD (C4CMMRD). Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium 'care for CMMRD' (C4CMMRD). J Med Genet. 2014 Jun;51(6):355-65. doi: 10.1136/jmedgenet-2014-102284. Epub 2014 Apr 15. Citation on PubMed
- Wimmer K, Rosenbaum T, Messiaen L. Connections between constitutional mismatch repair deficiency syndrome and neurofibromatosis type 1. Clin Genet. 2017 Apr;91(4):507-519. doi: 10.1111/cge.12904. Epub 2017 Jan 10. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.