

Description
Charcot-Marie-Tooth disease is a group of disorders this is characterized by damage to the nerves that are involved in muscle movement and sensation (motor and sensory neuropathy). This condition mainly affects peripheral nerves, which connect the brain and spinal cord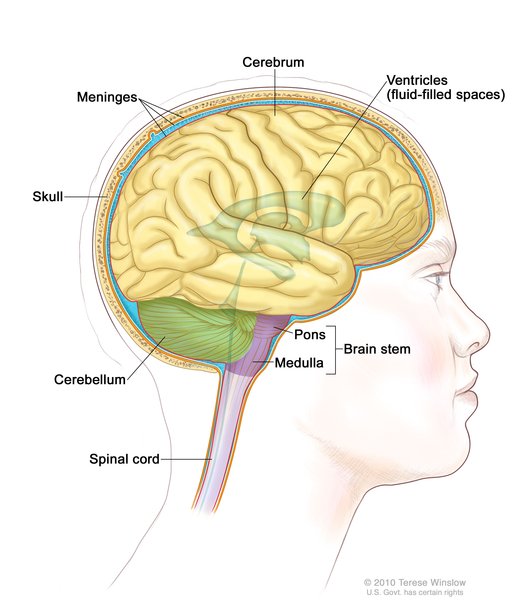 to muscles and to sensory cells that detect sensations such as touch, pain, and heat. In people with Charcot-Marie-Tooth disease, damage to the peripheral nerves worsens over time and can cause changes in sensation and wasting (atrophy) of muscles in the feet, legs, and hands.
to muscles and to sensory cells that detect sensations such as touch, pain, and heat. In people with Charcot-Marie-Tooth disease, damage to the peripheral nerves worsens over time and can cause changes in sensation and wasting (atrophy) of muscles in the feet, legs, and hands.
The severity of Charcot-Marie-Tooth disease varies widely. Some people with this condition have mild symptoms, but many develop progressive muscle weakness and sensory loss that can lead to physical disability and difficulty with balance, walking, and other daily activities. In a small percentage of affected individuals, weakness becomes severe or complications develop that can significantly affect health and, in rare cases, may be life-threatening. However, most people with Charcot-Marie-Tooth disease typically have a normal life expectancy.
Charcot-Marie-Tooth disease usually becomes apparent in childhood or adolescence, but the signs and symptoms may begin any time between early childhood and late adulthood. Typically, the earliest symptoms of Charcot-Marie-Tooth disease are caused by muscle weakness and atrophy in the feet. Affected individuals may have foot abnormalities such as high arches (pes cavus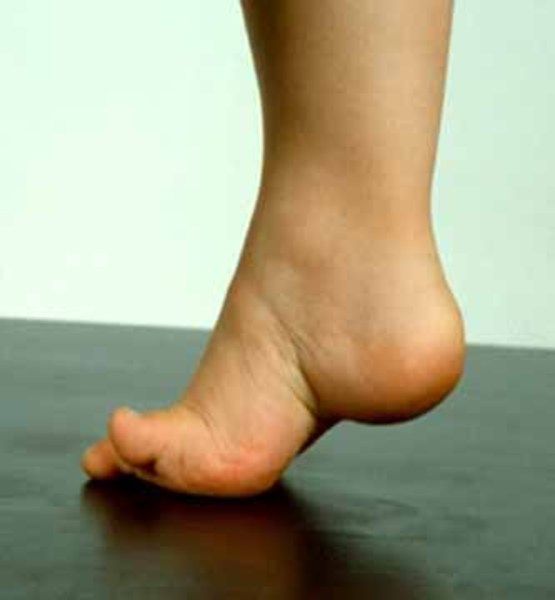 ), flat feet (pes planus
), flat feet (pes planus ), or curled toes (hammer toes). People with Charcot-Marie-Tooth disease often have difficulty flexing their foot or walking on the heel of the foot. This may make it difficult to lift the front part of the foot (foot drop) or cause a higher-than-normal step when walking (steppage gait), and people with this condition have a higher risk of ankle injuries and tripping. Younger children with this condition may have delayed development of motor skills such as walking. As the disease worsens, muscles in the lower legs usually weaken, which can affect walking and balance. Affected individuals often use supportive devices such as braces or orthotics to improve mobility; however, they rarely need wheelchair assistance.
), or curled toes (hammer toes). People with Charcot-Marie-Tooth disease often have difficulty flexing their foot or walking on the heel of the foot. This may make it difficult to lift the front part of the foot (foot drop) or cause a higher-than-normal step when walking (steppage gait), and people with this condition have a higher risk of ankle injuries and tripping. Younger children with this condition may have delayed development of motor skills such as walking. As the disease worsens, muscles in the lower legs usually weaken, which can affect walking and balance. Affected individuals often use supportive devices such as braces or orthotics to improve mobility; however, they rarely need wheelchair assistance.
People with Charcot-Marie-Tooth disease may also develop weakness in the hands, which causes difficulty with daily activities such as writing, fastening buttons, and turning doorknobs. Affected individuals typically experience a decreased sensitivity to touch, heat, and cold in the feet and lower legs, but they many occasionally experience aching, burning or other types of nerve pain. In rare cases, affected individuals have hearing or vision loss.
There are several types of Charcot-Marie-Tooth disease, and they are differentiated by their effects on nerve cells and their pattern of inheritance. The types are sometimes simply identified by the specific gene involved. These are the main types of Charcot-Marie-Tooth disease:
- Type 1 (CMT1) accounts for at least half of all cases of Charcot-Marie-Tooth disease. CMT1 is characterized by abnormalities in myelin, an insulating layer formed by specialized cells that wrap around nerve fibers. Normally, myelin helps nerve signals travel efficiently, but problems with myelin can slow the transmission of nerve signals.
- Type 2 (CMT2) is characterized by damage to part of the nerve called an axon. Axons extend from nerve cells and transmit nerve impulses between the brain, spinal cord, and muscles. Damage to axons reduces the strength of nerve signals. CMT2 accounts for 10 to 15 percent of all cases of Charcot-Marie-Tooth disease.
- Type 3 (CMT3), formerly known as Dejerine-Sottas syndrome (DSS), is a severe early-onset form of Charcot-Marie-Tooth disease that typically appears in infancy. This form affects myelin.
- Type 4 (CMT4) typically has a different pattern of inheritance than the other types; it can affect either the axons or myelin. CMT4 is rare and accounts for a small percentage of all cases of Charcot-Marie-Tooth disease.
- Type X Charcot-Marie-Tooth disease (CMTX) can cause abnormalities in the axons and myelin. CMTX accounts for 10 to 15 percent of all cases of Charcot-Marie-Tooth disease.
Within the various types of Charcot-Marie-Tooth disease, subtypes (such as CMT1A, CMT1B, CMT2A, CMT4A, and CMTX1) indicate different genetic causes. Some forms of Charcot-Marie-Tooth disease are classified as intermediate type. In people with these types, the nerve impulses are both slowed and reduced in strength, likely due to abnormalities in both myelin and axons.
Like DSS, other historical names are sometimes used to describe certain forms of Charcot-Marie-Tooth disease. For example, Roussy-Levy syndrome refers to a form of CMT1 that includes rhythmic shaking (tremor), and Rosenberg-Chutorian syndrome is a rare type of CMTX.
Frequency
Charcot-Marie-Tooth disease is the most common inherited disorder that involves the peripheral nerves, affecting an estimated 125,000 people in the United States. It occurs in populations worldwide, with a prevalence of about 1 in 2,500 individuals.
Causes
Genetic changes that cause disease are called pathogenic variants. Pathogenic variants in dozens of different genes can cause Charcot-Marie-Tooth disease. These genes provide instructions for making proteins that are involved in the function of peripheral nerves. The variants that are associated with Charcot-Marie-Tooth disease can cause cells to produce proteins that do not work properly. These changes affect myelin or the axons of nerve cells and slow down or weaken nerve signals. Longer nerves that run from the spinal cord to the hands and feet are especially vulnerable. As a result, peripheral nerve cells slowly lose the ability to stimulate the muscles in the feet, legs, and hands, and to transmit sensory signals from these areas to the brain. Different variants in the same gene can lead to different types of Charcot-Marie-Tooth disease.
Pathogenic variants in the PMP22, MPZ, MFN2, and GJB1 genes account for 80 to 90 percent of the genetic causes of Charcot-Marie-Tooth disease. Most people with CMT1 have variants that affect the PMP22 gene. These cases typically occur when there is an extra copy of the gene due to a small piece genetic material on chromosome 17 being abnormally copied (duplicated). Another 5 to 10 percent of individuals with CMT1 have variants in the MPZ gene. Variants in the MFN2 gene are the most common cause of CMT2, while approximately 90 percent of people with CMTX have GJB1 gene variants.
Pathogenic variants in many other genes have been identified in smaller numbers of individuals with Charcot-Marie-Tooth disease. As research advances, the number of genes associated with Charcot-Marie-Tooth disease continues to grow.
Inheritance
The pattern of inheritance varies depending on the type of Charcot-Marie-Tooth disease.
CMT1, most cases of CMT2, and most intermediate forms are inherited in an autosomal dominant pattern, which means that one copy of the altered gene in each cell is sufficient to cause the disorder.
which means that one copy of the altered gene in each cell is sufficient to cause the disorder.
CMT4, a few CMT2 subtypes, and some intermediate forms are inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. In autosomal recessive inheritance, the parents of an affected individual each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. In autosomal recessive inheritance, the parents of an affected individual each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
CMTX is inherited in an X-linked pattern. A condition is considered X-linked if the altered gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes. In males (who have only one X chromosome), a pathogenic variant in the only copy of the gene in each cell is sufficient to cause the condition. A characteristic of X-linked inheritance is that fathers cannot pass X-linked variants to their sons. In females (who have two copies of the X chromosome), one altered copy of the gene generally causes milder signs and symptoms of the disorder than those seen in affected males. However, in some cases, females with CMTX may have signs and symptoms that are as severe as those in males.
In males (who have only one X chromosome), a pathogenic variant in the only copy of the gene in each cell is sufficient to cause the condition. A characteristic of X-linked inheritance is that fathers cannot pass X-linked variants to their sons. In females (who have two copies of the X chromosome), one altered copy of the gene generally causes milder signs and symptoms of the disorder than those seen in affected males. However, in some cases, females with CMTX may have signs and symptoms that are as severe as those in males.
About 10 percent of cases of Charcot-Marie-Tooth disease result from a new (de novo) variant in the gene that occurs during the formation of reproductive cells (eggs or sperm) in an affected individual's parent or during early embryonic development. These affected individuals typically have no history of the disorder in their family.
Other Names for This Condition
- Charcot-Marie-Tooth hereditary neuropathy
- Charcot-Marie-Tooth syndrome
- CMT
- Hereditary motor and sensory neuropathy
- HMSN
- Inherited peripheral neuropathy
- Peroneal muscular atrophy
- PMA
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- CHARCOT-MARIE-TOOTH DISEASE AND DEAFNESS
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, AUTOSOMAL DOMINANT, TYPE 2A2A; CMT2A2A
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, AUTOSOMAL RECESSIVE, TYPE 2A2B; CMT2A2B
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, TYPE 2I; CMT2I
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, TYPE 2J; CMT2J
- CHARCOT-MARIE-TOOTH DISEASE, DEMYELINATING, TYPE 1A; CMT1A
- CHARCOT-MARIE-TOOTH DISEASE, DEMYELINATING, TYPE 1B; CMT1B
- CHARCOT-MARIE-TOOTH DISEASE, DOMINANT INTERMEDIATE D; CMTDID
- CHARCOT-MARIE-TOOTH DISEASE, X-LINKED DOMINANT, 1; CMTX1
- HYPERTROPHIC NEUROPATHY OF DEJERINE-SOTTAS
- ROUSSY-LEVY HEREDITARY AREFLEXIC DYSTASIA
Scientific Articles on PubMed
References
- Ando M, Higuchi Y, Yuan J, Yoshimura A, Taniguchi T, Takei J, Takeuchi M, Hiramatsu Y, Shimizu F, Kubota M, Takeshima A, Ueda T, Koh K, Nagaoka U, Tokashiki T, Sawai S, Sakiyama Y, Hashiguchi A, Sato R, Kanda T, Okamoto Y, Takashima H. Novel heterozygous variants of SLC12A6 in Japanese families with Charcot-Marie-Tooth disease. Ann Clin Transl Neurol. 2022 Jul;9(7):902-911. doi: 10.1002/acn3.51603. Epub 2022 Jun 22. Citation on PubMed
- Barreto LC, Oliveira FS, Nunes PS, de Franca Costa IM, Garcez CA, Goes GM, Neves EL, de Souza Siqueira Quintans J, de Souza Araujo AA. Epidemiologic Study of Charcot-Marie-Tooth Disease: A Systematic Review. Neuroepidemiology. 2016;46(3):157-65. doi: 10.1159/000443706. Epub 2016 Feb 6. Citation on PubMed
- Berciano J, Garcia A, Gallardo E, Peeters K, Pelayo-Negro AL, Alvarez-Paradelo S, Gazulla J, Martinez-Tames M, Infante J, Jordanova A. Intermediate Charcot-Marie-Tooth disease: an electrophysiological reappraisal and systematic review. J Neurol. 2017 Aug;264(8):1655-1677. doi: 10.1007/s00415-017-8474-3. Epub 2017 Mar 31. Citation on PubMed
- Bird TD. Charcot-Marie-Tooth Hereditary Neuropathy Overview. 1998 Sep 28 [updated 2025 Nov 20]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1358/ Citation on PubMed
- Deenen JC, Verbeek AL, Verschuuren JJ, van Engelen BG, Voermans NC. Prevalence and incidence rates of 17 neuromuscular disorders: An updated review of the literature. J Neuromuscul Dis. 2025 Nov;12(6):713-722. doi: 10.1177/22143602241313118. Epub 2025 Mar 4. Citation on PubMed
- El-Abassi R, England JD, Carter GT. Charcot-Marie-Tooth disease: an overview of genotypes, phenotypes, and clinical management strategies. PM R. 2014 Apr;6(4):342-55. doi: 10.1016/j.pmrj.2013.08.611. Epub 2014 Jan 13. Citation on PubMed
- Fridman V, Saporta MA. Mechanisms and Treatments in Demyelinating CMT. Neurotherapeutics. 2021 Oct;18(4):2236-2268. doi: 10.1007/s13311-021-01145-z. Epub 2021 Nov 8. Citation on PubMed
- Gutmann L, Shy M. Update on Charcot-Marie-Tooth disease. Curr Opin Neurol. 2015 Oct;28(5):462-7. doi: 10.1097/WCO.0000000000000237. Citation on PubMed
- Hoyle JC, Isfort MC, Roggenbuck J, Arnold WD. The genetics of Charcot-Marie-Tooth disease: current trends and future implications for diagnosis and management. Appl Clin Genet. 2015 Oct 19;8:235-43. doi: 10.2147/TACG.S69969. eCollection 2015. Citation on PubMed or Free article on PubMed Central
- Jani-Acsadi A, Ounpuu S, Pierz K, Acsadi G. Pediatric Charcot-Marie-Tooth disease. Pediatr Clin North Am. 2015 Jun;62(3):767-86. doi: 10.1016/j.pcl.2015.03.012. Epub 2015 Apr 15. Citation on PubMed
- Kazamel M, Boes CJ. Charcot Marie Tooth disease (CMT): historical perspectives and evolution. J Neurol. 2015;262(4):801-5. doi: 10.1007/s00415-014-7490-9. Epub 2014 Sep 9. Citation on PubMed
- Moss KR, Bopp TS, Johnson AE, Hoke A. New evidence for secondary axonal degeneration in demyelinating neuropathies. Neurosci Lett. 2021 Jan 23;744:135595. doi: 10.1016/j.neulet.2020.135595. Epub 2020 Dec 24. Citation on PubMed
- Wang Y, Yin F. A Review of X-linked Charcot-Marie-Tooth Disease. J Child Neurol. 2016 May;31(6):761-72. doi: 10.1177/0883073815604227. Epub 2015 Sep 18. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.