

Description
Aicardi syndrome is a rare disorder that occurs primarily in women and girls. Aicardi syndrome has typically been characterized by the presence of three main features:
- Absent or underdeveloped tissue that connects the left and right halves of the brain (agenesis or dysgenesis of the corpus callosum
 )
) - Defects in the retina, which is the light-sensitive tissue at the back of the eye (chorioretinal lacunae)
- Seizures that develop within the first year of life (infantile spasms)
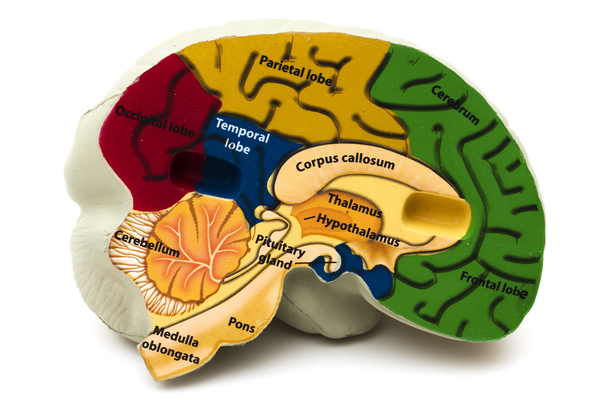
Although many affected individuals will have all three of these features, some people with Aicardi syndrome will have only two. Besides these main features, people with Aicardi syndrome typically have additional signs and symptoms.
The folds and grooves on the surface of the brain often do not develop properly in people with Aicardi syndrome. Additional abnormalities that can occur in affected individuals include a difference in the size or shape between the two halves of the brain, cysts in the brain, enlargement of the fluid-filled cavities (ventricles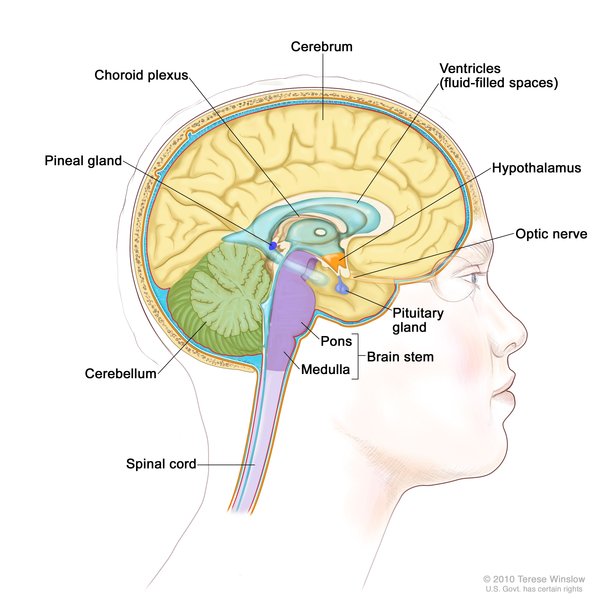 ) near the center of the brain, and clusters of cells that are not in the correct location (heterotopias).
) near the center of the brain, and clusters of cells that are not in the correct location (heterotopias).
Infantile spasms are the most common type of seizure in infants with Aicardi syndrome. However, as they age, many people with Aicardi syndrome will develop additional types of seizures, some of which may not respond well to medication.
People with Aicardi syndrome typically have developmental delays and intellectual disabilities, which can range from mild to severe. For most affected individuals, developmental delays and intellectual disabilities fall within the moderate to severe range.
Chorioretinal lacunae are considered to be a characteristic feature of Aicardi syndrome. However, additional eye abnormalities may also occur. The structure that carries information from the eye to the brain (optic nerve ) can be underdeveloped (hypoplasia) or have a gap or hole (coloboma). Another eye abnormality that can occur is called microphthalmia, which is a birth defect in which one or both eyes do not fully develop and are abnormally small. The eye abnormalities in individuals with Aicardi syndrome can cause vision loss.
) can be underdeveloped (hypoplasia) or have a gap or hole (coloboma). Another eye abnormality that can occur is called microphthalmia, which is a birth defect in which one or both eyes do not fully develop and are abnormally small. The eye abnormalities in individuals with Aicardi syndrome can cause vision loss.
Some people with Aicardi syndrome have differences between the right and left sides of the face (facial asymmetry), a short area between the upper lip and the nose (philtrum), a flat nose with an upturned tip, large ears, and sparse eyebrows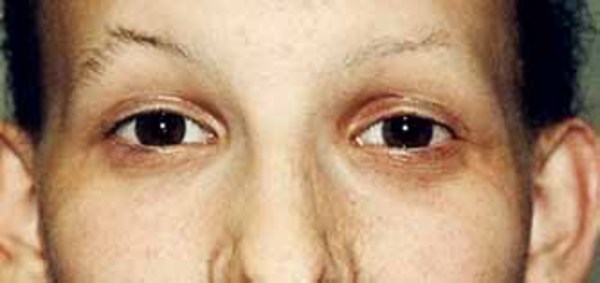 . Other features of this condition include small or malformed hands; spinal and rib abnormalities that lead to progressive abnormal curvature of the spine (scoliosis
. Other features of this condition include small or malformed hands; spinal and rib abnormalities that lead to progressive abnormal curvature of the spine (scoliosis ); and gastrointestinal problems, such as constipation or diarrhea, gastroesophageal reflux, and difficulty feeding.
); and gastrointestinal problems, such as constipation or diarrhea, gastroesophageal reflux, and difficulty feeding.
The number and severity of the signs and symptoms seen in people with Aicardi syndrome can vary. Affected individuals with severe signs and symptoms may not survive past childhood, while those with milder features can survive into adulthood.
Frequency
Aicardi syndrome is a very rare disorder. It occurs in about 1 in 105,000 to 167,000 newborns in the United States. Researchers estimate that there are approximately 4,000 affected individuals worldwide.
Causes
The cause of Aicardi syndrome is unknown. Because it occurs almost exclusively in women and girls, researchers believe that Aicardi syndrome is caused by changes in a gene on the X chromosome. Genetic changes that cause disease are called pathogenic variants.
People normally have 46 chromosomes in each cell. Two of the 46 chromosomes, known as X and Y, are called sex chromosomes because they help determine whether a person will develop male or female sex characteristics. Females typically have two X chromosomes (46,XX), and males have one X chromosome and one Y chromosome (46,XY).
because they help determine whether a person will develop male or female sex characteristics. Females typically have two X chromosomes (46,XX), and males have one X chromosome and one Y chromosome (46,XY).
Early in embryonic development in females, one of the two X chromosomes is permanently inactivated in cells other than egg and sperm cells. This process is called X-inactivation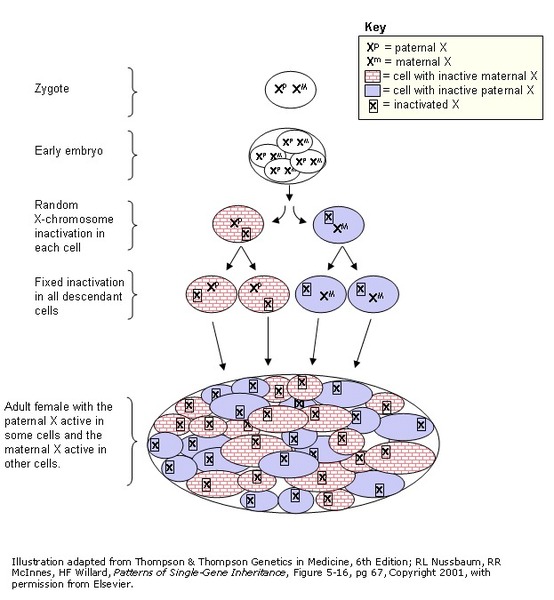 . X-inactivation ensures that females, like males, have only one active copy of the X chromosome in each body cell. X-inactivation usually occurs randomly, so that each X chromosome is active in about half the body's cells. Sometimes, however, X-inactivation is not random. This is called skewed X-inactivation.
. X-inactivation ensures that females, like males, have only one active copy of the X chromosome in each body cell. X-inactivation usually occurs randomly, so that each X chromosome is active in about half the body's cells. Sometimes, however, X-inactivation is not random. This is called skewed X-inactivation.
Skewed X-inactivation sometimes occurs when there is a pathogenic variant in one of the X chromosomes in each cell. Skewed X-inactivation has been identified in some women and girls with Aicardi syndrome. This could explain why some people with Aicardi syndrome are more severely affected than others. Skewed X inactivation also supports the idea that the condition is caused by a pathogenic variant in a gene on the X chromosome. However, the gene that contains this variant has not been identified.
Inheritance
Nearly all known cases of Aicardi syndrome are sporadic, which means that they occur in people with no history of the disorder in their family.
Researchers suspect that the pathogenic variants that cause Aicardi syndrome typically occur as random (de novo) events during the formation of eggs or sperm in an affected individual's parent or during early embryonic development.
Although this condition primarily affects women and girls, cases of men and boys with Aicardi syndrome have been reported.
Other Names for This Condition
- Agenesis of corpus callosum with chorioretinal abnormality
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Aicardi J. Aicardi syndrome. Brain Dev. 2005 Apr;27(3):164-71. doi: 10.1016/j.braindev.2003.11.011. Citation on PubMed
- Anderson S, Menten B, Kogelenberg Mv, Robertson S, Waginger M, Mentzel HJ, Brandl U, Skirl G, Willems P. Aicardi syndrome in a male patient. Neuropediatrics. 2009 Feb;40(1):39-42. doi: 10.1055/s-0029-1220760. Epub 2009 Jul 28. Citation on PubMed
- Chappelow AV, Reid J, Parikh S, Traboulsi EI. Aicardi syndrome in a genotypic male. Ophthalmic Genet. 2008 Dec;29(4):181-3. doi: 10.1080/13816810802320209. Citation on PubMed
- Eble TN, Sutton VR, Sangi-Haghpeykar H, Wang X, Jin W, Lewis RA, Fang P, Van den Veyver IB. Non-random X chromosome inactivation in Aicardi syndrome. Hum Genet. 2009 Mar;125(2):211-6. doi: 10.1007/s00439-008-0615-4. Epub 2009 Jan 1. Citation on PubMed or Free article on PubMed Central
- Glasmacher MA, Sutton VR, Hopkins B, Eble T, Lewis RA, Park Parsons D, Van den Veyver IB. Phenotype and management of Aicardi syndrome: new findings from a survey of 69 children. J Child Neurol. 2007 Feb;22(2):176-84. doi: 10.1177/0883073807300298. Citation on PubMed
- Grosso S, Lasorella G, Russo A, Galluzzi P, Morgese G, Balestri P. Aicardi syndrome with favorable outcome: case report and review. Brain Dev. 2007 Aug;29(7):443-6. doi: 10.1016/j.braindev.2006.11.011. Epub 2007 Jan 4. Citation on PubMed
- Kroner BL, Preiss LR, Ardini MA, Gaillard WD. New incidence, prevalence, and survival of Aicardi syndrome from 408 cases. J Child Neurol. 2008 May;23(5):531-5. doi: 10.1177/0883073807309782. Epub 2008 Jan 8. Citation on PubMed
- Masnada S, De Giorgis V, Carugo U, Bahi-Buisson N, Cavallin M, Corbett M, Formica M, Gecz J, Petros N, Perucca E, Pichiecchio A, Fusar Poli P, Sherr EH, Van den Veyver IB, Zara F, Geroldinger M, Veggiotti P, Arzimanoglou A; Aicardi Syndrome Study Group. Refining Aicardi Syndrome diagnostic Criteria: an expert-based consensus using a modified Delphi approach. Eur J Paediatr Neurol. 2026 Jan;60:58-70. doi: 10.1016/j.ejpn.2025.11.004. Epub 2025 Nov 27. Citation on PubMed
- Sutton VR, Hopkins BJ, Eble TN, Gambhir N, Lewis RA, Van den Veyver IB. Facial and physical features of Aicardi syndrome: infants to teenagers. Am J Med Genet A. 2005 Oct 15;138A(3):254-8. doi: 10.1002/ajmg.a.30963. Citation on PubMed
- Sutton VR, Van den Veyver IB. Aicardi Syndrome. 2006 Jun 30 [updated 2020 Nov 12]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1381/ Citation on PubMed
- Tuft M, Ostby Y, Nakken KO, Lund C. Aicardi syndrome and cognitive abilities: A report of five cases. Epilepsy Behav. 2017 Aug;73:161-165. doi: 10.1016/j.yebeh.2017.05.002. Epub 2017 Jul 18. Citation on PubMed
- Wong BKY, Sutton VR. Aicardi syndrome, an unsolved mystery: Review of diagnostic features, previous attempts, and future opportunities for genetic examination. Am J Med Genet C Semin Med Genet. 2018 Dec;178(4):423-431. doi: 10.1002/ajmg.c.31658. Epub 2018 Dec 10. Citation on PubMed
- Zubairi MS, Carter RF, Ronen GM. A male phenotype with Aicardi syndrome. J Child Neurol. 2009 Feb;24(2):204-7. doi: 10.1177/0883073808322337. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.