

Description
Yuan-Harel-Lupski (YUHAL) syndrome is a rare neurological condition that has a combination of features of two other disorders, Potocki-Lupski syndrome and type 1A Charcot-Marie-Tooth disease.
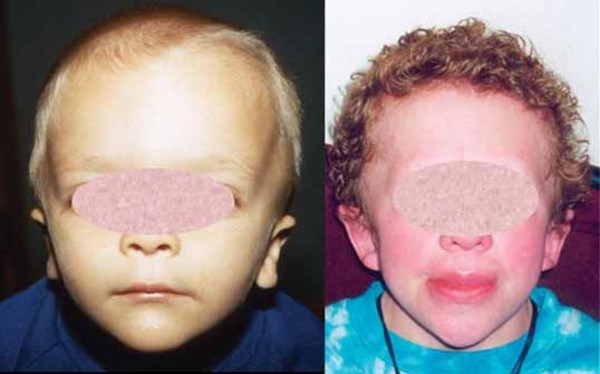
The first signs and symptoms of YUHAL syndrome begin in infancy. Infants with YUHAL syndrome usually have weak muscle tone (hypotonia), which may lead to feeding problems. They typically do not grow and gain weight at the expected rate. Babies and children with YUHAL syndrome have delayed development, including delayed speech and language skills and motor skills such as walking. YUHAL syndrome is also associated with behavioral difficulties. Many affected individuals have sleep problems, including pauses in breathing during sleep (sleep apnea) or trouble falling asleep and staying asleep. Some people with YUHAL syndrome have subtle differences in facial features, including outside corners of the eyes that point downward (down-slanting palpebral fissures), a triangular face, and eyes that do not look in the same direction (strabismus). These signs and symptoms are similar to those of Potocki-Lupski syndrome.
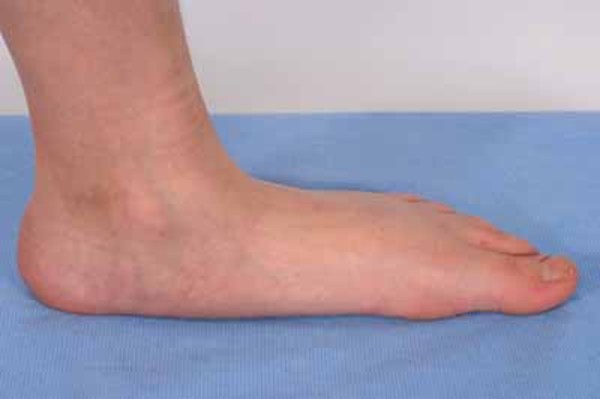
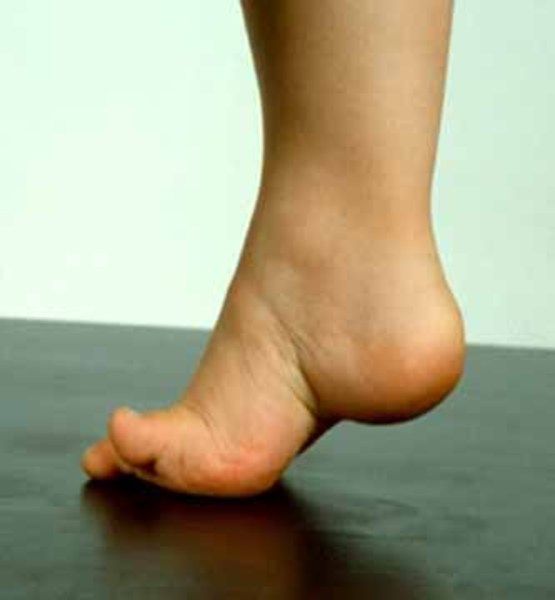
Other signs and symptoms of YUHAL syndrome begin in childhood and result from damage to peripheral nerves, which connect the brain and spinal cord to muscles and to sensory cells that detect sensations such as touch, pain, and heat. Damage to peripheral nerves can lead to loss of sensation and wasting (atrophy) of muscles in the legs. Children with YUHAL syndrome often develop muscle weakness, particularly in the lower legs, which may lead to an unusual walking style (gait). Some affected individuals have foot abnormalities such as flat feet (pes planus), high arches (pes cavus), or an inward- and upward-turning foot (clubfoot). They may also experience reduced reflexes and a decreased sensitivity to touch, heat, and cold in the feet and lower legs. Similar features are seen in individuals with type 1A Charcot-Marie-Tooth disease, although they may appear earlier in people with YUHAL syndrome, often before age 5.

Abnormal development of other tissues and organs, such as the heart or kidneys, can occur in YUHAL syndrome.
Frequency
The prevalence of YUHAL syndrome is unknown. More than 20 people with the condition have been described in the medical literature.
Causes
YUHAL syndrome results from an extra copy (duplication) of a small piece of chromosome 17 in each cell. The duplication occurs on the short (p) arm of the chromosome in a region designated 17p12-17p11.2. The duplicated segment ranges in size from approximately 3.2 million DNA building blocks (also written as 3.2 megabases or 3.2 Mb) to 19.7 Mb.
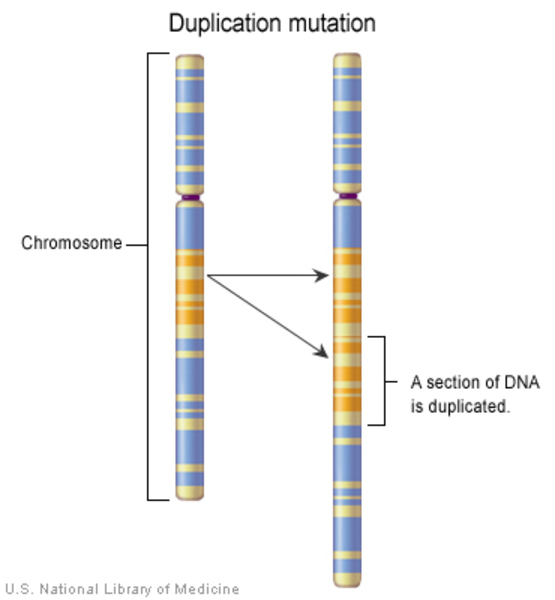
The duplicated region always contains at least two genes, RAI1 and PMP22. Researchers believe that having an extra copy of both of these genes underlies the characteristic features of YUHAL syndrome.
The RAI1 gene provides instructions for making a protein that helps regulate the activity (expression) of other genes. Although most of the genes regulated by the RAI1 protein have not been identified, the protein appears to play a role in many body processes, including the sleep-wake cycle and development of the brain and bones in the head and face (craniofacial bones). Research suggests that duplications involving this gene lead to higher-than-normal amounts of the RAI1 protein, which disrupts the expression of genes that influence brain and craniofacial development and the sleep-wake cycle. Excess RAI1 protein likely causes feeding and sleep problems, delayed development, behavioral difficulties, and facial differences in people with YUHAL syndrome. Duplications that involve the RAI1 gene but not the PMP22 gene cause Potocki-Lupski syndrome, which shares these features.
The PMP22 gene provides instructions for making a protein that is a component of myelin, a protective substance that covers nerves and promotes the efficient transmission of nerve impulses. It is thought that an extra copy of the PMP22 gene leads to overproduction of PMP22 protein. Too much PMP22 protein may overwhelm the cells' ability to process it correctly, leading to a buildup of unprocessed, nonfunctional protein. This buildup may impair the formation of myelin. A shortage of functional PMP22 protein leads to instability and loss of myelin (demyelination). Demyelination reduces the ability of the peripheral nerves to activate muscles used for movement or to relay information from sensory cells back to the brain. As a result, individuals with a duplication of the PMP22 gene develop muscle weakness and impaired sensitivity to touch, heat, and cold. Duplications that involve the PMP22 gene but not the RAI1 gene cause type 1A Charcot-Marie-Tooth disease, which is characterized by similar problems with muscle weakness and sensation.

Researchers suspect that having an extra copy of additional genes in the duplicated region contribute to other features of the disorder, such as kidney abnormalities.
Inheritance
YUHAL syndrome follows an autosomal dominant pattern of inheritance, which means one copy of the chromosome duplication in each cell is sufficient to cause the disorder. Most cases of this condition result from new (de novo) duplications that occur during the formation of reproductive cells (eggs or sperm) or in early embryonic development. These cases occur in people with no history of the disorder in their family.

Other Names for This Condition
- PMP22-RAI1 contiguous gene duplication syndrome
- YUHAL syndrome
Additional Information & Resources
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Doco-Fenzy M, Holder-Espinasse M, Bieth E, Magdelaine C, Vincent MC, Khoury M, Andrieux J, Zhang F, Lupski JR, Klink R, Schneider A, Goze-Martineau O, Cuisset JM, Vallee L, Manouvrier-Hanu S, Gaillard D, de Martinville B. The clinical spectrum associated with a chromosome 17 short arm proximal duplication (dup 17p11.2) in three patients. Am J Med Genet A. 2008 Apr 1;146A(7):917-24. doi: 10.1002/ajmg.a.32195. Citation on PubMed
- Shaw CJ, Stankiewicz P, Christodoulou J, Smith E, Jones K, Lupski JR. A girl with duplication 17p10-p12 associated with a dicentric chromosome. Am J Med Genet A. 2004 Jan 15;124A(2):173-8. doi: 10.1002/ajmg.a.20355. Citation on PubMed
- Yuan B, Harel T, Gu S, Liu P, Burglen L, Chantot-Bastaraud S, Gelowani V, Beck CR, Carvalho CM, Cheung SW, Coe A, Malan V, Munnich A, Magoulas PL, Potocki L, Lupski JR. Nonrecurrent 17p11.2p12 Rearrangement Events that Result in Two Concomitant Genomic Disorders: The PMP22-RAI1 Contiguous Gene Duplication Syndrome. Am J Hum Genet. 2015 Nov 5;97(5):691-707. doi: 10.1016/j.ajhg.2015.10.003. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.