

Description
Treacher Collins syndrome is a condition that affects the development of bones and other tissues of the face. The signs and symptoms of this disorder vary greatly, ranging from almost unnoticeable to severe. Most affected individuals have underdeveloped facial bones, particularly the cheek bones, and a very small jaw and chin (micrognathia). Some people with this condition are also born with an opening in the roof of the mouth called a cleft palate . In severe cases, underdevelopment of the facial bones may restrict an affected infant's airway, causing potentially life-threatening respiratory problems.
. In severe cases, underdevelopment of the facial bones may restrict an affected infant's airway, causing potentially life-threatening respiratory problems.
People with Treacher Collins syndrome often have eyes that slant downward, sparse eyelashes, and a notch in the lower eyelids called an eyelid coloboma. Some affected individuals have additional eye abnormalities that can lead to vision loss. This condition is also characterized by absent, small, or unusually formed ears . Hearing loss occurs in about half of all affected individuals; hearing loss is caused by defects of the three small bones in the middle ear, which transmit sound, or by underdevelopment of the ear canal. People with Treacher Collins syndrome usually have normal intelligence.
. Hearing loss occurs in about half of all affected individuals; hearing loss is caused by defects of the three small bones in the middle ear, which transmit sound, or by underdevelopment of the ear canal. People with Treacher Collins syndrome usually have normal intelligence.
Frequency
This condition affects an estimated 1 in 50,000 people.
Causes
Variants (also known as mutations) in the TCOF1, POLR1C, or POLR1D gene can cause Treacher Collins syndrome. TCOF1 gene variants are the most common cause of the disorder, accounting for 81 to 93 percent of all cases. POLR1C and POLR1D gene variants cause an additional 2 percent of cases. In individuals without an identified variant in one of these genes, the genetic cause of the condition is unknown.
The proteins produced from the TCOF1, POLR1C, and POLR1D genes all appear to play important roles in the early development of bones and other tissues of the face. These proteins are involved in the production of a molecule called ribosomal RNA (rRNA), a chemical cousin of DNA. Ribosomal RNA helps assemble protein building blocks (amino acids) into new proteins, which is essential for the normal functioning and survival of cells.
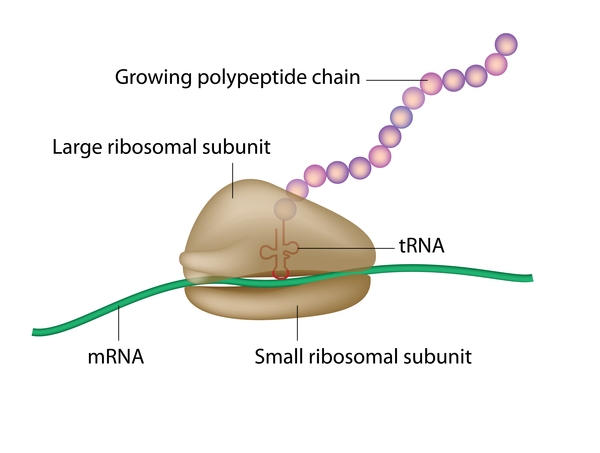
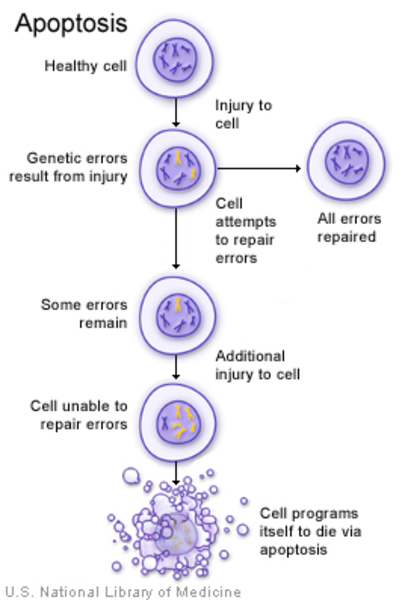
Variants in the TCOF1, POLR1C, or POLR1D gene reduce the production of rRNA. Researchers speculate that a decrease in the amount of rRNA may trigger the self-destruction (apoptosis) of certain cells involved in the development of facial bones and tissues. The abnormal cell death could lead to the specific problems with facial development found in Treacher Collins syndrome. However, it is unclear why the effects of a reduction in rRNA are limited to facial development.
Inheritance
When Treacher Collins syndrome results from variants in the TCOF1 or POLR1D gene, it is considered an autosomal dominant condition, which means one copy of the altered gene in each cell is sufficient to cause the disorder. About 60 percent of these cases result from new variants in the gene and occur in people with no history of the disorder in their family. In the remaining autosomal dominant cases, a person with Treacher Collins syndrome inherits the altered gene from an affected parent
in the gene and occur in people with no history of the disorder in their family. In the remaining autosomal dominant cases, a person with Treacher Collins syndrome inherits the altered gene from an affected parent .
.
When Treacher Collins syndrome is caused by variants in the POLR1C gene, the condition has an autosomal recessive pattern of inheritance. Autosomal recessive inheritance means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
pattern of inheritance. Autosomal recessive inheritance means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Franceschetti-Zwahlen-Klein syndrome
- Mandibulofacial dysostosis (MFD1)
- Treacher Collins-Franceschetti syndrome
- Zygoauromandibular dysplasia
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Barbosa M, Jabs EW, Huston S. Treacher Collins Syndrome. 2004 Jul 20 [updated 2024 Jun 20]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1532/ Citation on PubMed
- Dauwerse JG, Dixon J, Seland S, Ruivenkamp CA, van Haeringen A, Hoefsloot LH, Peters DJ, Boers AC, Daumer-Haas C, Maiwald R, Zweier C, Kerr B, Cobo AM, Toral JF, Hoogeboom AJ, Lohmann DR, Hehr U, Dixon MJ, Breuning MH, Wieczorek D. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat Genet. 2011 Jan;43(1):20-2. doi: 10.1038/ng.724. Epub 2010 Dec 5. Citation on PubMed
- Dixon J, Jones NC, Sandell LL, Jayasinghe SM, Crane J, Rey JP, Dixon MJ, Trainor PA. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc Natl Acad Sci U S A. 2006 Sep 5;103(36):13403-8. doi: 10.1073/pnas.0603730103. Epub 2006 Aug 28. Citation on PubMed or Free article on PubMed Central
- Marszalek B, Wojcicki P, Kobus K, Trzeciak WH. Clinical features, treatment and genetic background of Treacher Collins syndrome. J Appl Genet. 2002;43(2):223-33. Citation on PubMed
- Posnick JC, Ruiz RL. Treacher Collins syndrome: current evaluation, treatment, and future directions. Cleft Palate Craniofac J. 2000 Sep;37(5):434. doi: 10.1597/1545-1569(2000)0372.0.CO;2. Citation on PubMed
- Sakai D, Trainor PA. Treacher Collins syndrome: unmasking the role of Tcof1/treacle. Int J Biochem Cell Biol. 2009 Jun;41(6):1229-32. doi: 10.1016/j.biocel.2008.10.026. Epub 2008 Nov 5. Citation on PubMed or Free article on PubMed Central
- Teber OA, Gillessen-Kaesbach G, Fischer S, Bohringer S, Albrecht B, Albert A, Arslan-Kirchner M, Haan E, Hagedorn-Greiwe M, Hammans C, Henn W, Hinkel GK, Konig R, Kunstmann E, Kunze J, Neumann LM, Prott EC, Rauch A, Rott HD, Seidel H, Spranger S, Sprengel M, Zoll B, Lohmann DR, Wieczorek D. Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation. Eur J Hum Genet. 2004 Nov;12(11):879-90. doi: 10.1038/sj.ejhg.5201260. Citation on PubMed
- Trainor PA, Dixon J, Dixon MJ. Treacher Collins syndrome: etiology, pathogenesis and prevention. Eur J Hum Genet. 2009 Mar;17(3):275-83. doi: 10.1038/ejhg.2008.221. Epub 2008 Dec 24. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.