

Description
Sturge-Weber syndrome is a condition that affects the development of certain blood vessels, causing abnormalities in the brain, skin, and eyes from birth. Sturge-Weber syndrome has three major features: a red or pink birthmark called a port-wine birthmark, a brain abnormality called a leptomeningeal angioma, and increased pressure in the eye (glaucoma). These features can vary in severity and not all individuals with Sturge-Weber syndrome have all three features.
Most people with Sturge-Weber syndrome are born with a port-wine birthmark. This type of birthmark is caused by enlargement (dilatation) of small blood vessels (capillaries) near the surface of the skin. Port-wine birthmarks are typically initially flat and can vary in color from pale pink to deep purple. In people with Sturge-Weber syndrome, the port-wine birthmark is most often on the face, typically on the forehead, temple, or eyelid. The port-wine birthmark is usually only on one side of the face but can be on both sides. Over time, the skin within the port-wine birthmark can darken and thicken.
In Sturge-Weber syndrome, there is usually abnormal formation and growth of blood vessels within the two thin layers of tissue that cover the brain and spinal cord. This abnormality, which is called leptomeningeal angioma, can affect one or both sides of the brain and impair blood flow in the brain and lead to loss of brain tissue (atrophy) and deposits of calcium (calcification) in the brain below the angioma. The decrease in blood flow caused by leptomeningeal angiomas can cause stroke-like episodes in people with Sturge-Weber syndrome. These episodes often involve temporary muscle weakness on one side of the body (hemiparesis), vision abnormalities, seizures, and migraine headaches. In affected individuals, these episodes usually begin by age 2. The seizures usually involve only one side of the brain (focal seizures), during which the port-wine birthmark may darken and individuals may lose consciousness. People with Sturge-Weber syndrome have varying levels of cognitive function, from normal intelligence to intellectual disability. Some individuals have learning disabilities with problems focusing similar to attention-deficit/hyperactivity disorder (ADHD).
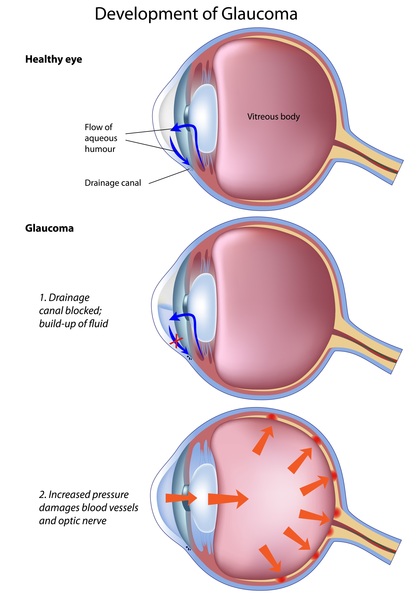
In individuals with Sturge-Weber syndrome, glaucoma typically develops either in infancy or early adulthood and can cause vision impairment. In some affected infants, the pressure can become so great that the eyeballs appear enlarged and bulging (buphthalmos). Individuals with Sturge-Weber syndrome can have tangles of abnormal blood vessels (hemangiomas) in various parts of the eye. When these abnormal blood vessels develop in the network of blood vessels at the back of the eye (choroid), it is called a diffuse choroidal hemangioma and occurs in about one-third of individuals with Sturge-Weber syndrome. A diffuse choroidal hemangioma can cause vision loss. When present, the eye abnormalities typically occur on the same side of the head as the port-wine birthmark.
Frequency
Sturge-Weber syndrome is estimated to affect 1 in 20,000 to 50,000 individuals.
Causes
Sturge-Weber syndrome is caused by a mutation in the GNAQ gene. This gene provides instructions for making a protein called guanine nucleotide-binding protein G(q) subunit alpha (Gαq). The Gαq protein is part of a group of proteins (complex) that regulates signaling pathways to help control the development and function of blood vessels.
The GNAQ gene mutation that causes Sturge-Weber syndrome results in the production of a protein with impaired function. As a result, the altered Gαq protein cannot play its part in regulating signaling pathways, resulting in abnormally increased signaling. The enhanced signaling likely disrupts the regulation of blood vessel development, causing abnormal and excessive formation of vessels before birth in people with Sturge-Weber syndrome.
Inheritance
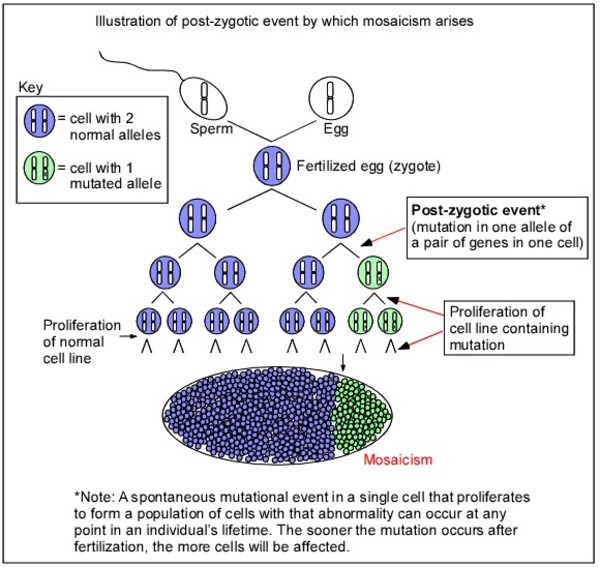
Sturge-Weber syndrome is not inherited. The mutation that causes this disorder is somatic, which means it occurs after conception. In Sturge-Weber syndrome, the mutation is thought to occur in a cell during early development before birth. As that cell continues to grow and divide, the cells derived from it, specifically certain cells in the brain, eyes, and skin that are involved in blood vessel formation, also have the mutation, while the body's other cells do not. This situation is called mosaicism. The mosaic nature of the mutations helps to explain why the abnormal blood vessel growth occurs in some parts of the body but not in others.
Other Names for This Condition
- Angiomatosis aculoorbital-thalamic syndrome
- Encephalofacial hemangiomatosis
- Encephalofacial hemangiomatosis syndrome
- Meningo-oculo-facial angiomatosis
- Meningofacial angiomatosis-cerebral calcification syndrome
- Neuroretinoangiomatosis
- Phakomatosis, Sturge-Weber
- Sturge-Weber-Dimitri syndrome
- Sturge-Weber-Krabbe syndrome
- SWS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Comi AM, Sahin M, Hammill A, Kaplan EH, Juhasz C, North P, Ball KL, Levin AV, Cohen B, Morris J, Lo W, Roach ES; 2015 Sturge-Weber Syndrome Research Workshop. Leveraging a Sturge-Weber Gene Discovery: An Agenda for Future Research. Pediatr Neurol. 2016 May;58:12-24. doi: 10.1016/j.pediatrneurol.2015.11.009. Epub 2016 Mar 16. Citation on PubMed
- Comi AM. Sturge-Weber syndrome. Handb Clin Neurol. 2015;132:157-68. doi: 10.1016/B978-0-444-62702-5.00011-1. Citation on PubMed
- Koenraads Y, van Egmond-Ebbeling MB, de Boer JH, Imhof SM, Braun KP, Porro GL; SWS study group. Visual outcome in Sturge-Weber syndrome: a systematic review and Dutch multicentre cohort. Acta Ophthalmol. 2016 Nov;94(7):638-645. doi: 10.1111/aos.13074. Epub 2016 May 30. Citation on PubMed
- Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, North PE, Marchuk DA, Comi AM, Pevsner J. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013 May 23;368(21):1971-9. doi: 10.1056/NEJMoa1213507. Epub 2013 May 8. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.