

Description
Renal hypouricemia is a kidney (renal) disorder that results in a reduced amount of urate in the blood. Urate is a byproduct of certain normal chemical reactions in the body. In the bloodstream it acts as an antioxidant, protecting cells from the damaging effects of unstable molecules called free radicals. However, having too much urate in the body is toxic, so excess urate is removed from the body in urine.
People with renal hypouricemia have little to no urate in their blood; they release an excessive amount of it in the urine. In many affected individuals, renal hypouricemia causes no signs or symptoms. However, some people with this condition develop kidney problems. After strenuous exercise, they can develop exercise-induced acute kidney injury, which causes pain in their sides and lower back as well as nausea and vomiting that can last several hours.
Because an excessive amount of urate passes through the kidneys to be excreted in urine in people with renal hypouricemia, they have an increased risk of developing kidney stones (nephrolithiasis) formed from urate crystals. These urate stones can damage the kidneys and lead to episodes of blood in the urine (hematuria). Rarely, people with renal hypouricemia develop life-threatening kidney failure.
Frequency
The prevalence of renal hypouricemia is unknown; at least 150 affected individuals have been described in the scientific literature. This condition is thought to be most prevalent in Asian countries such as Japan and South Korea, although affected individuals have been found in Europe. Renal hypouricemia is likely underdiagnosed because it does not cause any symptoms in many affected individuals.
Causes
Mutations in the SLC22A12 or SLC2A9 gene cause renal hypouricemia. These genes provide instructions for making proteins called urate transporter 1 (URAT1) and glucose transporter 9 (GLUT9), respectively. These proteins are found in the kidneys, specifically in structures called proximal tubules. These structures help to reabsorb needed nutrients, water, and other materials into the blood and excrete unneeded substances into the urine. Within the proximal tubules, both the URAT1 and GLUT9 proteins reabsorb urate into the bloodstream or release it into the urine, depending on the body's needs. Most urate that is filtered through the kidneys is reabsorbed into the bloodstream; about 10 percent is released into urine.
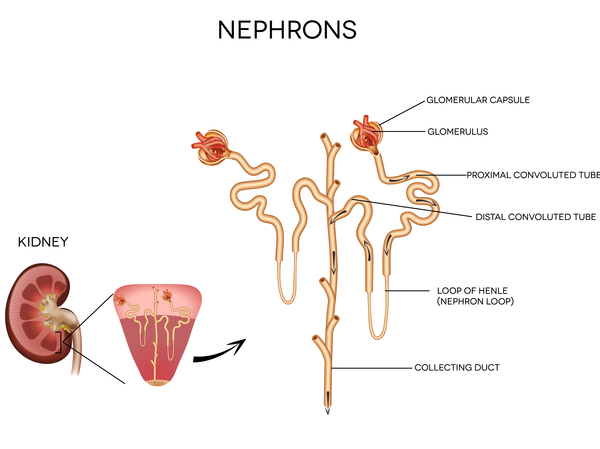
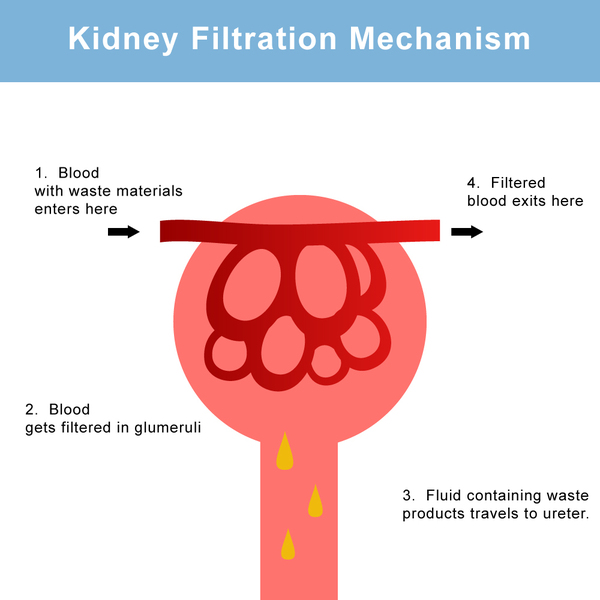
Mutations that cause renal hypouricemia lead to the production of URAT1 or GLUT9 protein with a reduced ability to reabsorb urate into the bloodstream. Instead, large amounts of urate are released in the urine. The specific cause of the signs and symptoms of renal hypouricemia is unclear. Researchers suspect that when additional urate is produced during exercise and passed through the kidneys, it could lead to tissue damage. Alternatively, without the antioxidant properties of urate, free radicals could cause tissue damage in the kidneys. Another possibility is that other substances are prevented from being reabsorbed along with urate; accumulation of these substances in the kidneys could cause tissue damage. It is likely that individuals with renal hypouricemia who have mild or no symptoms have enough protein function to reabsorb a sufficient amount of urate into the bloodstream to prevent severe kidney problems.
Inheritance
This condition is typically inherited in an autosomal recessive pattern, which means both copies of the SLC22A12 or SLC2A9 gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they usually do not show signs and symptoms of the condition.

Sometimes, individuals with one SLC2A9 gene mutation in each cell have reduced levels of urate. The levels usually are not as low as they are in people who have mutations in both copies of the gene, and they often do not cause any signs or symptoms. Rarely, people who carry one copy of the mutated gene will develop urate kidney stones.
Other Names for This Condition
- Familial renal hypouricaemia
- Familial renal hypouricemia
- Hereditary renal hypouricemia
- RHUC
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Kaito H, Ishimori S, Nozu K, Shima Y, Nakanishi K, Yoshikawa N, Iijima K. Molecular background of urate transporter genes in patients with exercise-induced acute kidney injury. Am J Nephrol. 2013;38(4):316-20. doi: 10.1159/000355430. Epub 2013 Oct 4. Citation on PubMed
- Kawamura Y, Matsuo H, Chiba T, Nagamori S, Nakayama A, Inoue H, Utsumi Y, Oda T, Nishiyama J, Kanai Y, Shinomiya N. Pathogenic GLUT9 mutations causing renal hypouricemia type 2 (RHUC2). Nucleosides Nucleotides Nucleic Acids. 2011 Dec;30(12):1105-11. doi: 10.1080/15257770.2011.623685. Citation on PubMed
- Stiburkova B, Sebesta I, Ichida K, Nakamura M, Hulkova H, Krylov V, Kryspinova L, Jahnova H. Novel allelic variants and evidence for a prevalent mutation in URAT1 causing renal hypouricemia: biochemical, genetics and functional analysis. Eur J Hum Genet. 2013 Oct;21(10):1067-73. doi: 10.1038/ejhg.2013.3. Epub 2013 Feb 6. Citation on PubMed or Free article on PubMed Central
- Tasic V, Hynes AM, Kitamura K, Cheong HI, Lozanovski VJ, Gucev Z, Jutabha P, Anzai N, Sayer JA. Clinical and functional characterization of URAT1 variants. PLoS One. 2011;6(12):e28641. doi: 10.1371/journal.pone.0028641. Epub 2011 Dec 16. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.