

Description
Renal coloboma syndrome (also known as papillorenal syndrome) is a condition that primarily affects kidney (renal) and eye development. People with this condition typically have kidneys that are small and underdeveloped (hypoplastic), which can lead to end-stage renal disease (ESRD). This serious disease occurs when the kidneys are no longer able to filter fluids and waste products from the body effectively. It has been estimated that approximately ten percent of children with hypoplastic kidneys may have renal coloboma syndrome. The kidney problems can affect one or both kidneys.
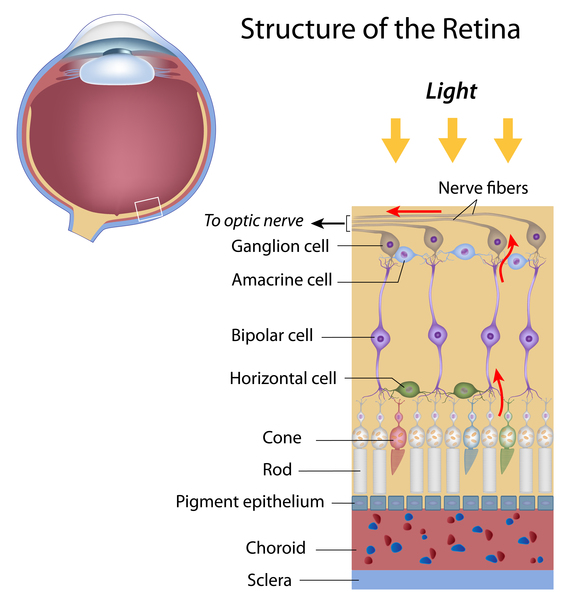
Additionally, people with renal coloboma syndrome may have a malformation in the optic nerve, a structure that carries information from the eye to the brain. Optic nerve malformations are sometimes associated with a gap or hole (coloboma) in the light-sensitive tissue at the back of the eye (the retina). The vision problems caused by these abnormalities can vary depending on the size and location of the malformation. Some people have no visual problems, while others may have severely impaired vision.
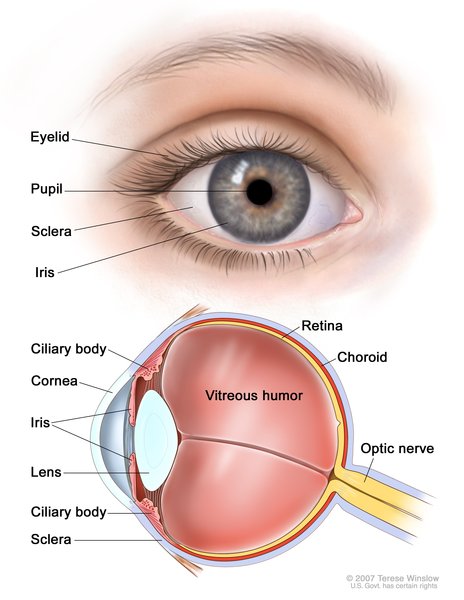
Less common features of renal coloboma syndrome include backflow of urine from the bladder (vesicoureteral reflux), multiple kidney cysts, loose joints, and mild hearing loss.
Frequency
The prevalence of renal coloboma syndrome is unknown; at least 60 cases have been reported in the scientific literature.
Causes
Renal coloboma syndrome is caused by variants (also known as mutations) in the PAX2 gene. The PAX2 gene provides instructions for making a protein that is involved in the early development of the eyes, ears, brain and spinal cord (central nervous system), kidneys , and genital tract. The PAX2 protein attaches (binds) to specific regions of DNA and regulates the activity of other genes. On the basis of this role, the PAX2 protein is called a transcription factor. After birth, the PAX2 protein is thought to protect against cell death during periods of cellular stress.
, and genital tract. The PAX2 protein attaches (binds) to specific regions of DNA and regulates the activity of other genes. On the basis of this role, the PAX2 protein is called a transcription factor. After birth, the PAX2 protein is thought to protect against cell death during periods of cellular stress.
Variants in the PAX2 gene lead to the production of a nonfunctional PAX2 protein that is unable to aid in development, causing incomplete formation of certain tissues. Why the kidneys and eyes are specifically affected by PAX2 gene variants is unclear.
Approximately half of those affected with renal coloboma syndrome do not have an identified variant in the PAX2 gene. In these cases, the cause of the disorder is unknown.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Other Names for This Condition
- Coloboma of optic nerve with renal disease
- Coloboma-ureteral-renal syndrome
- ONCR
- Optic coloboma, vesicoureteral reflux, and renal anomalies
- Optic nerve coloboma renal syndrome
- Papillorenal syndrome
- RCS
- Renal-coloboma syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Amiel J, Audollent S, Joly D, Dureau P, Salomon R, Tellier AL, Auge J, Bouissou F, Antignac C, Gubler MC, Eccles MR, Munnich A, Vekemans M, Lyonnet S, Attie-Bitach T. PAX2 mutations in renal-coloboma syndrome: mutational hotspot and germline mosaicism. Eur J Hum Genet. 2000 Nov;8(11):820-6. doi: 10.1038/sj.ejhg.5200539. Citation on PubMed
- Bower MA, Schimmenti LA, Eccles MR. PAX2-Related Disorder. 2007 Jun 8 [updated 2025 Aug 14]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1451/ Citation on PubMed
- Cheong HI, Cho HY, Kim JH, Yu YS, Ha IS, Choi Y. A clinico-genetic study of renal coloboma syndrome in children. Pediatr Nephrol. 2007 Sep;22(9):1283-9. doi: 10.1007/s00467-007-0525-z. Epub 2007 May 31. Citation on PubMed
- Dureau P, Attie-Bitach T, Salomon R, Bettembourg O, Amiel J, Uteza Y, Dufier JL. Renal coloboma syndrome. Ophthalmology. 2001 Oct;108(10):1912-6. doi: 10.1016/s0161-6420(01)00722-9. Citation on PubMed
- Schimmenti LA, Manligas GS, Sieving PA. Optic nerve dysplasia and renal insufficiency in a family with a novel PAX2 mutation, Arg115X: further ophthalmologic delineation of the renal-coloboma syndrome. Ophthalmic Genet. 2003 Dec;24(4):191-202. doi: 10.1076/opge.24.4.191.17229. Citation on PubMed
- Weber S, Moriniere V, Knuppel T, Charbit M, Dusek J, Ghiggeri GM, Jankauskiene A, Mir S, Montini G, Peco-Antic A, Wuhl E, Zurowska AM, Mehls O, Antignac C, Schaefer F, Salomon R. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol. 2006 Oct;17(10):2864-70. doi: 10.1681/ASN.2006030277. Epub 2006 Sep 13. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.