

Description
Neurodevelopmental disorder with or without anomalies of the brain, eye, or heart (NEDBEH) is a neurological disorder that can also affect many other body systems. This condition primarily affects neurological development, causing intellectual disability, delayed development of speech and motor skills (such as sitting and walking), or autism spectrum disorder, which is a condition that affects communication and social interaction. Some affected individuals have additional neurological features, such as weak muscle tone (hypotonia), behavioral problems, and seizures.
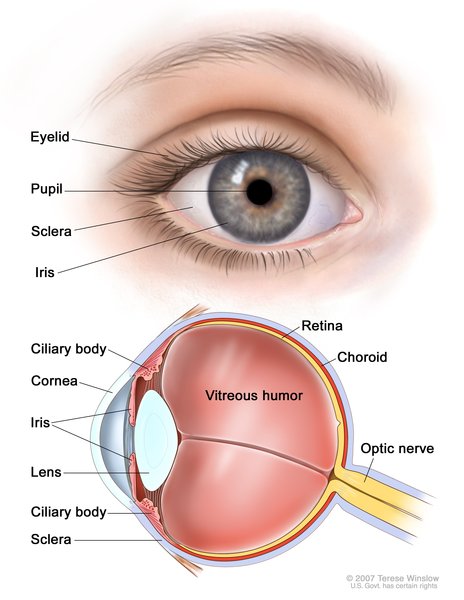
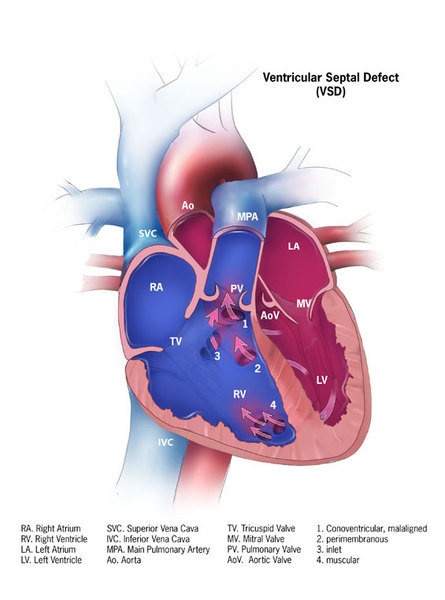
NEDBEH can affect development of many other parts of the body. Some affected individuals have abnormalities of brain structures, such as the tissue that connects the left and right halves of the brain (the corpus callosum), a tissue called white matter, the fluid-filled cavities (ventricles) near the center of the brain, or a structure at the back of the brain known as the cerebellar vermis. Eye abnormalities that can occur include a gap or hole in one of the structures of the eye (coloboma), underdevelopment (hypoplasia) or breakdown (atrophy) of the nerves that carry information from the eyes to the brain (optic nerves), or unusually small eyeballs (microphthalmia). These eye problems can cause vision impairment. Some affected individuals have heart defects, most commonly ventricular septal defect, which is a hole in the muscular wall (septum) that separates the right and left sides of the heart's lower chambers.

Less commonly, other systems are affected in NEDBEH, including the kidneys and inner ear. Problems with the inner ear can lead to hearing impairment (sensorineural hearing loss).

The signs and symptoms in some people with NEDBEH resemble those of another condition known as CHARGE syndrome; however, people with NEDBEH do not have changes in the gene associated with CHARGE syndrome.
Frequency
NEDBEH is a rare disorder with unknown prevalence. About twenty individuals with this condition have been described in the medical literature.
Causes
NEDBEH is caused by mutations in a gene called RERE. This gene provides instructions for making a protein that is critical for normal development before birth. It helps control the activity of a number of genes that are important for early development of the brain, eyes, inner ear, heart, and kidneys.
Researchers suspect that RERE gene mutations reduce or eliminate the function of the RERE protein. A shortage of RERE protein function likely alters the activity of several genes involved in development before birth. These changes prevent the normal development of tissues in the brain, eyes, heart, and other organs. Researchers are working to identify which genes are affected and how changes in their activity lead to the signs and symptoms of NEDBEH. It is unknown why some people with NEDBEH have only neurological problems and others also have structural abnormalities. Researchers suspect that the severity of the condition may be related to the location and type of mutation in the RERE gene. Additional genetic factors that have not been identified, including variations in other genes, may also help determine which body systems are affected.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Most cases of this condition result from new (de novo) mutations in the gene that occur during the formation of reproductive cells (eggs or sperm) in an affected individual’s parent or in early embryonic development. These cases occur in people with no history of the disorder in their family.
Other Names for This Condition
- NEDBEH
- RERE-related neurodevelopmental syndrome
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Fregeau B, Kim BJ, Hernandez-Garcia A, Jordan VK, Cho MT, Schnur RE, Monaghan KG, Juusola J, Rosenfeld JA, Bhoj E, Zackai EH, Sacharow S, Baranano K, Bosch DGM, de Vries BBA, Lindstrom K, Schroeder A, James P, Kulch P, Lalani SR, van Haelst MM, van Gassen KLI, van Binsbergen E, Barkovich AJ, Scott DA, Sherr EH. De Novo Mutations of RERE Cause a Genetic Syndrome with Features that Overlap Those Associated with Proximal 1p36 Deletions. Am J Hum Genet. 2016 May 5;98(5):963-970. doi: 10.1016/j.ajhg.2016.03.002. Epub 2016 Apr 14. Citation on PubMed or Free article on PubMed Central
- Jordan VK, Fregeau B, Ge X, Giordano J, Wapner RJ, Balci TB, Carter MT, Bernat JA, Moccia AN, Srivastava A, Martin DM, Bielas SL, Pappas J, Svoboda MD, Rio M, Boddaert N, Cantagrel V, Lewis AM, Scaglia F; Undiagnosed Diseases Network; Kohler JN, Bernstein JA, Dries AM, Rosenfeld JA, DeFilippo C, Thorson W, Yang Y, Sherr EH, Bi W, Scott DA. Genotype-phenotype correlations in individuals with pathogenic RERE variants. Hum Mutat. 2018 May;39(5):666-675. doi: 10.1002/humu.23400. Epub 2018 Jan 25. Citation on PubMed or Free article on PubMed Central
- Kim BJ, Scott DA. Mouse model reveals the role of RERE in cerebellar foliation and the migration and maturation of Purkinje cells. PLoS One. 2014 Jan 23;9(1):e87518. doi: 10.1371/journal.pone.0087518. eCollection 2014. Citation on PubMed or Free article on PubMed Central
- Kim BJ, Zaveri HP, Jordan VK, Hernandez-Garcia A, Jacob DJ, Zamora DL, Yu W, Schwartz RJ, Scott DA. RERE deficiency leads to decreased expression of GATA4 and the development of ventricular septal defects. Dis Model Mech. 2018 Aug 28;11(9):dmm031534. doi: 10.1242/dmm.031534. Citation on PubMed
- Kim BJ, Zaveri HP, Shchelochkov OA, Yu Z, Hernandez-Garcia A, Seymour ML, Oghalai JS, Pereira FA, Stockton DW, Justice MJ, Lee B, Scott DA. An allelic series of mice reveals a role for RERE in the development of multiple organs affected in chromosome 1p36 deletions. PLoS One. 2013;8(2):e57460. doi: 10.1371/journal.pone.0057460. Epub 2013 Feb 25. Citation on PubMed or Free article on PubMed Central
- Vilhais-Neto GC, Maruhashi M, Smith KT, Vasseur-Cognet M, Peterson AS, Workman JL, Pourquie O. Rere controls retinoic acid signalling and somite bilateral symmetry. Nature. 2010 Feb 18;463(7283):953-7. doi: 10.1038/nature08763. Citation on PubMed
- Zoltewicz JS, Stewart NJ, Leung R, Peterson AS. Atrophin 2 recruits histone deacetylase and is required for the function of multiple signaling centers during mouse embryogenesis. Development. 2004 Jan;131(1):3-14. doi: 10.1242/dev.00908. Epub 2003 Nov 26. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.