

Description
CHARGE syndrome is a disorder that affects many areas of the body. CHARGE is an abbreviation for several of the features common in the disorder: coloboma, heart defects, atresia choanae (also known as choanal atresia), growth retardation, genital abnormalities, and ear abnormalities. The pattern of malformations varies among individuals with this disorder, and the multiple health problems can be life-threatening in infancy. Affected individuals usually have several major characteristics or a combination of major and minor characteristics.
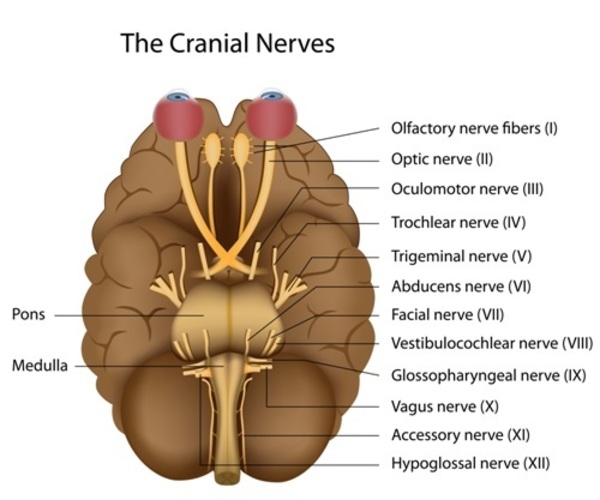
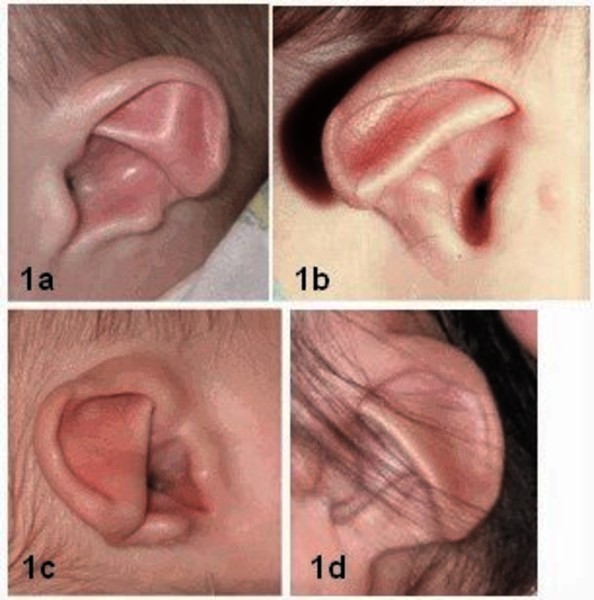
The major characteristics of CHARGE syndrome are common in this disorder and occur less frequently in other disorders. Most individuals with CHARGE syndrome have a gap or hole in one of the structures of the eye (coloboma), which forms during early development. A coloboma may be present in one or both eyes and may impair a person's vision, depending on its size and location. Some affected individuals also have abnormally small or underdeveloped eyes (microphthalmia). In many people with CHARGE syndrome, one or both nasal passages are narrowed (choanal stenosis) or completely blocked (choanal atresia), which can cause difficulty breathing. Affected individuals frequently have cranial nerve abnormalities. The cranial nerves emerge directly from the brain and extend to various areas of the head and neck, controlling muscle movement and transmitting sensory information. Abnormal function of certain cranial nerves can cause swallowing problems, facial paralysis, a sense of smell that is diminished (hyposmia) or completely absent (anosmia), and mild to profound hearing loss. People with CHARGE syndrome also typically have middle and inner ear abnormalities, which can contribute to hearing problems, and unusually shaped external ears.

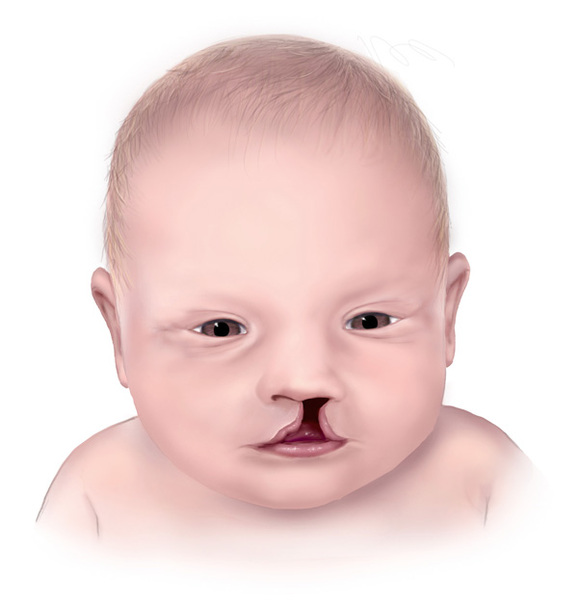
While the minor characteristics of CHARGE syndrome are common in this disorder, they are also frequently present in people without the disorder. The minor characteristics include heart defects; slow growth starting in late infancy; delayed development of motor skills, such as sitting unsupported and walking; and an opening in the lip (cleft lip) with or without an opening in the roof of the mouth (cleft palate). Affected individuals frequently have hypogonadotropic hypogonadism, which affects the production of hormones that direct sexual development. As a result, males with CHARGE syndrome are often born with an unusually small penis (micropenis) and undescended testes (cryptorchidism). Abnormalities of external genitalia are seen less often in affected females. Puberty can be incomplete or delayed in affected males and females. Another minor feature of CHARGE syndrome is tracheoesophageal fistula, which is an abnormal connection (fistula) between the esophagus and the trachea. Most people with CHARGE syndrome also have distinctive facial features, including a square-shaped face and differences in appearance between the right and left sides of the face (facial asymmetry). Affected individuals have a wide range of cognitive function, from normal intelligence to major learning disabilities with absent speech and poor communication.

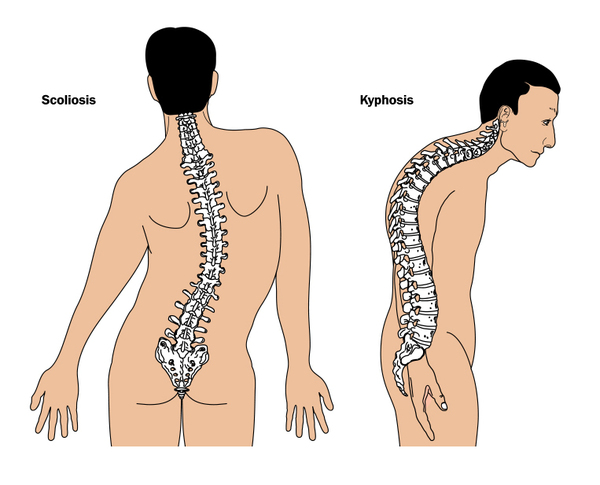
Less common features of CHARGE syndrome include kidney abnormalities; immune system problems; abnormal curvature of the spine (scoliosis or kyphosis); and limb abnormalities, such as extra fingers or toes (polydactyly), missing fingers or toes (oligodactyly), an inward and upward turning foot (club foot), and abnormalities of the long bones of the arms and legs.


Frequency
CHARGE syndrome occurs in approximately 1 in 8,500 to 10,000 newborns.
Causes
Mutations in the CHD7 gene cause most cases of CHARGE syndrome. The CHD7 gene provides instructions for making a protein that regulates gene activity (expression) by a process known as chromatin remodeling. Chromatin is the complex of DNA and protein that packages DNA into chromosomes. The structure of chromatin can be changed (remodeled) to alter how tightly DNA is packaged. When DNA is tightly packed, gene expression is lower than when DNA is loosely packed. Chromatin remodeling is one way gene expression is regulated during development.
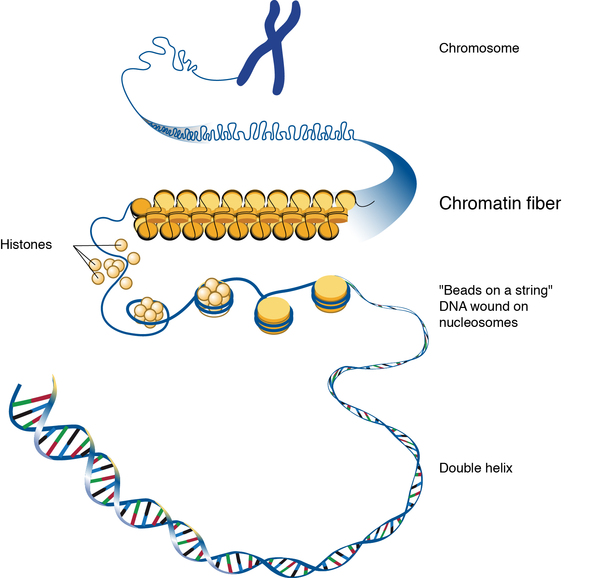
Most mutations in the CHD7 gene lead to the production of an abnormal CHD7 protein that is broken down prematurely. Shortage of this protein is thought to disrupt chromatin remodeling and the regulation of gene expression. Changes in gene expression during embryonic development likely cause the signs and symptoms of CHARGE syndrome.
A small percentage of individuals with CHARGE syndrome do not have an identified mutation in the CHD7 gene. Some of them may have a genetic change affecting the CHD7 gene that has not been found, and others may have a change in a different gene, although additional genes associated with CHARGE syndrome have not been identified.
Inheritance
When CHARGE syndrome is caused by mutations in the CHD7 gene, it follows an autosomal dominant pattern of inheritance, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Most cases result from new mutations in the gene and occur in people with no history of the disorder in their family. In rare cases, an affected person inherits the mutation from an affected parent. The inheritance pattern of other cases of CHARGE syndrome is unknown.


Other Names for This Condition
- CHARGE association
- Hall-Hittner syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bergman JE, Janssen N, Hoefsloot LH, Jongmans MC, Hofstra RM, van Ravenswaaij-Arts CM. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet. 2011 May;48(5):334-42. doi: 10.1136/jmg.2010.087106. Epub 2011 Mar 4. Citation on PubMed
- Blake KD, Prasad C. CHARGE syndrome. Orphanet J Rare Dis. 2006 Sep 7;1:34. doi: 10.1186/1750-1172-1-34. Citation on PubMed or Free article on PubMed Central
- Hale CL, Niederriter AN, Green GE, Martin DM. Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A. 2016 Feb;170A(2):344-354. doi: 10.1002/ajmg.a.37435. Epub 2015 Nov 21. Citation on PubMed or Free article on PubMed Central
- Lalani SR, Safiullah AM, Fernbach SD, Harutyunyan KG, Thaller C, Peterson LE, McPherson JD, Gibbs RA, White LD, Hefner M, Davenport SL, Graham JM, Bacino CA, Glass NL, Towbin JA, Craigen WJ, Neish SR, Lin AE, Belmont JW. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am J Hum Genet. 2006 Feb;78(2):303-14. doi: 10.1086/500273. Epub 2005 Dec 29. Citation on PubMed or Free article on PubMed Central
- Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clement-Ziza M, Delezoide AL, Aubry MC, Pelet A, Chemouny S, Cruaud C, Audollent S, Esculpavit C, Goudefroye G, Ozilou C, Fredouille C, Joye N, Morichon-Delvallez N, Dumez Y, Weissenbach J, Munnich A, Amiel J, Encha-Razavi F, Lyonnet S, Vekemans M, Attie-Bitach T. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet. 2006 Mar;43(3):211-217. doi: 10.1136/jmg.2005.036160. Epub 2005 Sep 16. Citation on PubMed or Free article on PubMed Central
- Sanlaville D, Verloes A. CHARGE syndrome: an update. Eur J Hum Genet. 2007 Apr;15(4):389-99. doi: 10.1038/sj.ejhg.5201778. Epub 2007 Feb 14. Citation on PubMed
- van Ravenswaaij-Arts CM, Hefner M, Blake K, Martin DM. CHD7 Disorder. 2006 Oct 2 [updated 2025 Aug 14]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1117/ Citation on PubMed
- Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A. 2005 Mar 15;133A(3):306-8. doi: 10.1002/ajmg.a.30559. No abstract available. Citation on PubMed
- Zentner GE, Layman WS, Martin DM, Scacheri PC. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet A. 2010 Mar;152A(3):674-86. doi: 10.1002/ajmg.a.33323. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.