

Description
Arginine vasopressin resistance (previously called nephrogenic diabetes insipidus) is a disorder of water balance. The body normally balances fluid intake by releasing excess fluid in urine. However, people with arginine vasopressin resistance produce an excessive amount of urine (polyuria), which depletes the amount of water in the body. This water loss also leads to excessive thirst (polydipsia).
Affected individuals can quickly become dehydrated if they do not drink enough water. Dehydration can cause dizziness and fatigue. Prolonged dehydration can lead to confusion, low blood pressure, seizures, and coma. People with arginine vasopressin resistance often develop high levels of sodium in the blood (hypernatremia) due to dehydration. Repeated cycles of dehydration can cause long-term health problems, particularly in children.
Arginine vasopressin resistance can be either acquired or familial. The acquired form can occur at any time during life. The familial form usually become apparent within the first year of life, though in some cases they develop in adolescence or early adulthood.
Infants with familial arginine vasopressin resistance tend to have problems feeding and gaining weight (failure to thrive). They may also be irritable and experience fevers, diarrhea, and vomiting. Recurrent episodes of dehydration can lead to slow growth and delayed development. If the condition is not well-managed, it can damage the bladder and kidneys leading to pain, infections, and kidney failure. With appropriate treatment, affected individuals usually have few complications and a normal lifespan.
Researchers have recommended using the condition name arginine vasopressin resistance because the previous name, nephrogenic diabetes insipidus, was often confused with a much more common disorder called diabetes mellitus.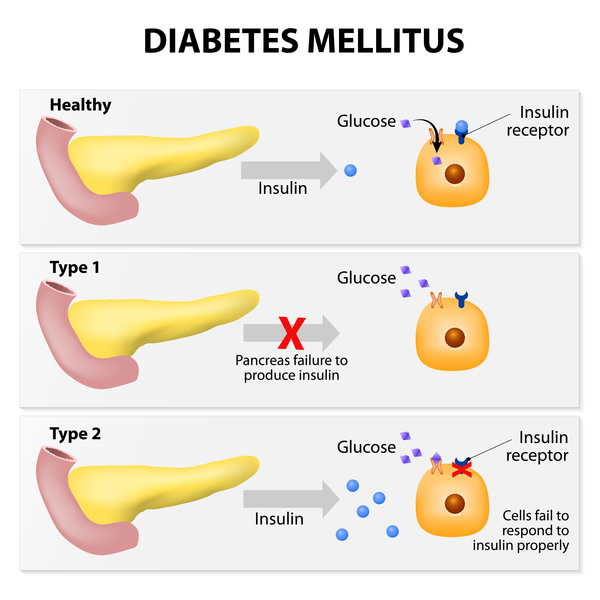 Arginine vasopressin resistance and diabetes mellitus are separate disorders with different features, causes, and treatment.
Arginine vasopressin resistance and diabetes mellitus are separate disorders with different features, causes, and treatment.
Frequency
The prevalence of arginine vasopressin resistance is unknown, although the condition is thought to be rare. The acquired form occurs more frequently than the familial form.
Causes
The familial form of arginine vasopressin resistance can be caused by variants (also called mutations) in at least two genes that are active in the kidneys. About 90 percent of all cases of familial arginine vasopressin resistance are caused by variants in the AVPR2 gene. Most of the remaining 10 percent of cases are caused by variants in the AQP2 gene. The AVPR2 gene provides instructions for making the vasopressin V2 receptor protein, and the AQP2 gene provides instructions for of the aquaporin-2 protein. Both of these proteins work with a hormone called arginine vasopressin (AVP) to help determine how much water is released in urine.
Normally, the kidneys filter the blood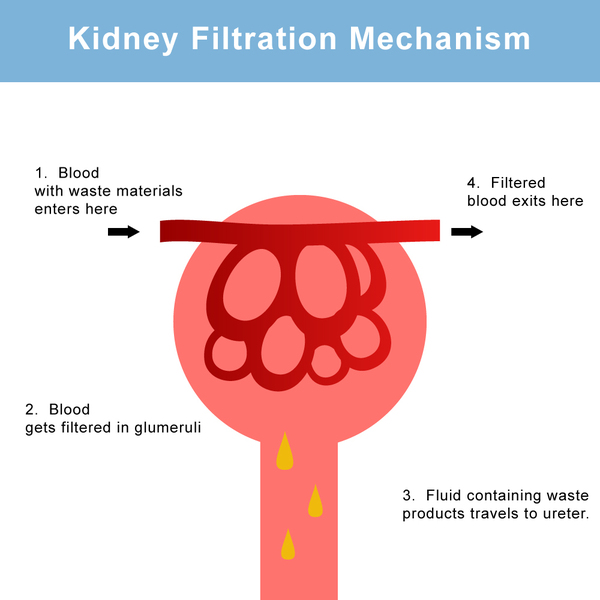 to remove waste and excess fluid, which are then stored in the bladder as urine. The balance between fluid intake and fluid release is controlled by AVP, which is sometimes also called antidiuretic hormone (ADH). AVP is produced and stored in the brain and it works with the vasopressin V2 receptor and aquaporin-2 proteins in the kidneys to manage fluid balance. Normally, when a person's fluid intake is low or when a lot of fluid is lost (for example, through sweating), the brain releases more AVP into the bloodstream. High levels of this hormone direct the kidneys to reabsorb more water and to make less urine. When fluid intake is adequate, the brain releases less AVP.
to remove waste and excess fluid, which are then stored in the bladder as urine. The balance between fluid intake and fluid release is controlled by AVP, which is sometimes also called antidiuretic hormone (ADH). AVP is produced and stored in the brain and it works with the vasopressin V2 receptor and aquaporin-2 proteins in the kidneys to manage fluid balance. Normally, when a person's fluid intake is low or when a lot of fluid is lost (for example, through sweating), the brain releases more AVP into the bloodstream. High levels of this hormone direct the kidneys to reabsorb more water and to make less urine. When fluid intake is adequate, the brain releases less AVP.
Variants in the AVPR2 or AQP2 genes lead to the production of proteins that are unable to respond to AVP. The brain produces and releases AVP normally, but the kidneys function as if little or no hormone is present. As a result, the kidneys do not reabsorb water as they should, and the body makes excessive amounts of urine. These problems with water balance are responsible for the dehydration that is characteristic of arginine vasopressin resistance.
The acquired form of arginine vasopressin resistance occurs when the brain is damaged due to head injuries, brain tumors, or other events. It can also be caused by chronic kidney disease, certain medications (such as lithium), low levels of potassium in the blood (hypokalemia), high levels of calcium in the blood (hypercalcemia), or an obstructed urinary tract. The acquired form of arginine vasopressin resistance also causes problems with water balance.
Inheritance
When arginine vasopressin resisitance is caused by variants in the AVPR2 gene, the condition has an X-linked pattern of inheritance. The AVPR2 gene is located on the X chromosome, which is one of the two sex chromosomes . In males (who have only one X chromosome), a variant in the only copy of the gene in each cell is sufficient to cause the condition. One altered copy of the gene can cause the condition in females (who have two copies of the X chromosome) as well, although the features may be less severe than those seen in individuals with two altered copies, or there may be no signs or symptoms at all. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
. In males (who have only one X chromosome), a variant in the only copy of the gene in each cell is sufficient to cause the condition. One altered copy of the gene can cause the condition in females (who have two copies of the X chromosome) as well, although the features may be less severe than those seen in individuals with two altered copies, or there may be no signs or symptoms at all. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
When arginine vasopressin resisitance is caused by variants in the AQP2 gene, it can have either an autosomal recessive or, less commonly, an autosomal dominant pattern of inheritance. In autosomal recessive inheritance , both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition. In autosomal dominant inheritance
, both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition. In autosomal dominant inheritance , one altered copy of the AQP2 gene in each cell is sufficient to cause the disorder.
, one altered copy of the AQP2 gene in each cell is sufficient to cause the disorder.
Other Names for This Condition
- ADH-resistant diabetes insipidus
- Congenital nephrogenic diabetes insipidus
- Diabetes insipidus renalis
- Diabetes insipidus, nephrogenic
- NDI
- Nephrogenic diabetes insipidus
- Vasopressin-resistant diabetes insipidus
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Atila C, Loughrey PB, Garrahy A, Winzeler B, Refardt J, Gildroy P, Hamza M, Pal A, Verbalis JG, Thompson CJ, Hemkens LG, Hunter SJ, Sherlock M, Levy MJ, Karavitaki N, Newell-Price J, Wass JAH, Christ-Crain M. Central diabetes insipidus from a patient's perspective: management, psychological co-morbidities, and renaming of the condition: results from an international web-based survey. Lancet Diabetes Endocrinol. 2022 Oct;10(10):700-709. doi: 10.1016/S2213-8587(22)00219-4. Epub 2022 Aug 22. Citation on PubMed
- Bichet DG. GENETICS IN ENDOCRINOLOGY Pathophysiology, diagnosis and treatment of familial nephrogenic diabetes insipidus. Eur J Endocrinol. 2020 Aug;183(2):R29-R40. doi: 10.1530/EJE-20-0114. Citation on PubMed
- Hureaux M, Vargas-Poussou R. Genetic basis of nephrogenic diabetes insipidus. Mol Cell Endocrinol. 2023 Jan 15;560:111825. doi: 10.1016/j.mce.2022.111825. Epub 2022 Nov 30. Citation on PubMed
- Khanna A. Acquired nephrogenic diabetes insipidus. Semin Nephrol. 2006 May;26(3):244-8. doi: 10.1016/j.semnephrol.2006.03.004. Citation on PubMed
- Knoers N, Lemmink H. Hereditary Nephrogenic Diabetes Insipidus. 2000 Feb 12 [updated 2020 Feb 27]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1177/ Citation on PubMed
- Knoers NV, Deen PM. Molecular and cellular defects in nephrogenic diabetes insipidus. Pediatr Nephrol. 2001 Dec;16(12):1146-52. doi: 10.1007/s004670100051. Citation on PubMed
- van Lieburg AF, Knoers NV, Monnens LA. Clinical presentation and follow-up of 30 patients with congenital nephrogenic diabetes insipidus. J Am Soc Nephrol. 1999 Sep;10(9):1958-64. doi: 10.1681/ASN.V1091958. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.