

Description
Mowat-Wilson syndrome is a genetic condition that affects many parts of the body. Major signs of this disorder frequently include distinctive facial features, intellectual disability, delayed development, an intestinal disorder called Hirschsprung disease, and other birth defects.
Children with Mowat-Wilson syndrome have a square-shaped face with deep-set, widely spaced eyes. They also have a broad nasal bridge with a rounded nasal tip; a prominent and pointed chin; large, flaring eyebrows; and uplifted earlobes with a dimple in the middle. These facial features become more distinctive with age, and adults with Mowat-Wilson syndrome have an elongated face with heavy eyebrows and a pronounced chin and jaw. Affected people tend to have a smiling, open-mouthed expression, and they typically have friendly and happy personalities.

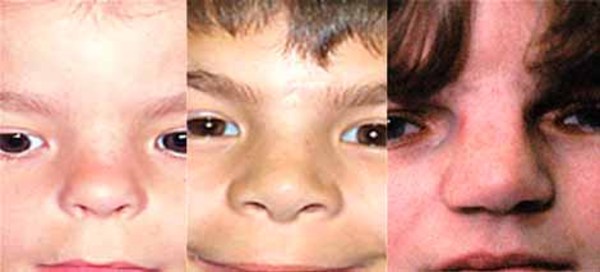
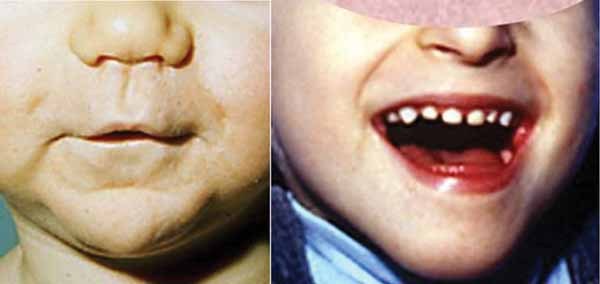
Mowat-Wilson syndrome is often associated with an unusually small head (microcephaly), structural brain abnormalities, and intellectual disability ranging from moderate to severe. Speech is absent or severely impaired, and affected people may learn to speak only a few words. Many people with this condition can understand others' speech, however, and some use sign language to communicate. If speech develops, it is delayed until mid-childhood or later. Children with Mowat-Wilson syndrome also have delayed development of motor skills such as sitting, standing, and walking.

More than half of people with Mowat-Wilson syndrome are born with an intestinal disorder called Hirschsprung disease that causes severe constipation, intestinal blockage, and enlargement of the colon. Chronic constipation also occurs frequently in people with Mowat-Wilson syndrome who have not been diagnosed with Hirschsprung disease.
Other features of Mowat-Wilson syndrome include short stature, seizures, heart defects, and abnormalities of the urinary tract and genitalia. Less commonly, this condition also affects the eyes, teeth, hands, and skin coloring (pigmentation). Although many different medical issues have been associated with Mowat-Wilson syndrome, not every individual with this condition has all of these features.

Frequency
The prevalence of Mowat-Wilson syndrome is unknown. More than 200 people with this condition have been reported in the medical literature.
Causes
Mutations in the ZEB2 gene cause Mowat-Wilson syndrome. The ZEB2 gene provides instructions for making a protein that plays a critical role in the formation of many organs and tissues before birth. This protein is a transcription factor, which means that it attaches (binds) to specific regions of DNA and helps control the activity of particular genes. Researchers believe that the ZEB2 protein is involved in the development of tissues that give rise to the nervous system, digestive tract, facial features, heart, and other organs.
Mowat-Wilson syndrome almost always results from a loss of one working copy of the ZEB2 gene in each cell. In some cases, the entire gene is deleted. In other cases, mutations within the gene lead to the production of an abnormally short, nonfunctional version of the ZEB2 protein. A shortage of this protein disrupts the normal development of many organs and tissues, which causes the varied signs and symptoms of Mowat-Wilson syndrome.

Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Other Names for This Condition
- Hirschsprung disease-mental retardation syndrome
- Microcephaly, mental retardation, and distinct facial features, with or without Hirschsprung disease
- MWS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Adam MP, Conta J, Bean LJH. Classic Mowat-Wilson Syndrome. 2007 Mar 28 [updated 2026 Apr 23]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1412/ Citation on PubMed
- Adam MP, Schelley S, Gallagher R, Brady AN, Barr K, Blumberg B, Shieh JT, Graham J, Slavotinek A, Martin M, Keppler-Noreuil K, Storm AL, Hudgins L. Clinical features and management issues in Mowat-Wilson syndrome. Am J Med Genet A. 2006 Dec 15;140(24):2730-41. doi: 10.1002/ajmg.a.31530. Citation on PubMed
- Evans E, Einfeld S, Mowat D, Taffe J, Tonge B, Wilson M. The behavioral phenotype of Mowat-Wilson syndrome. Am J Med Genet A. 2012 Feb;158A(2):358-66. doi: 10.1002/ajmg.a.34405. Epub 2012 Jan 13. Citation on PubMed
- Garavelli L, Zollino M, Mainardi PC, Gurrieri F, Rivieri F, Soli F, Verri R, Albertini E, Favaron E, Zignani M, Orteschi D, Bianchi P, Faravelli F, Forzano F, Seri M, Wischmeijer A, Turchetti D, Pompilii E, Gnoli M, Cocchi G, Mazzanti L, Bergamaschi R, De Brasi D, Sperandeo MP, Mari F, Uliana V, Mostardini R, Cecconi M, Grasso M, Sassi S, Sebastio G, Renieri A, Silengo M, Bernasconi S, Wakamatsu N, Neri G. Mowat-Wilson syndrome: facial phenotype changing with age: study of 19 Italian patients and review of the literature. Am J Med Genet A. 2009 Mar;149A(3):417-26. doi: 10.1002/ajmg.a.32693. Citation on PubMed
- Ishihara N, Yamada K, Yamada Y, Miura K, Kato J, Kuwabara N, Hara Y, Kobayashi Y, Hoshino K, Nomura Y, Mimaki M, Ohya K, Matsushima M, Nitta H, Tanaka K, Segawa M, Ohki T, Ezoe T, Kumagai T, Onuma A, Kuroda T, Yoneda M, Yamanaka T, Saeki M, Segawa M, Saji T, Nagaya M, Wakamatsu N. Clinical and molecular analysis of Mowat-Wilson syndrome associated with ZFHX1B mutations and deletions at 2q22-q24.1. J Med Genet. 2004 May;41(5):387-93. doi: 10.1136/jmg.2003.016154. No abstract available. Citation on PubMed or Free article on PubMed Central
- Mowat DR, Wilson MJ, Goossens M. Mowat-Wilson syndrome. J Med Genet. 2003 May;40(5):305-10. doi: 10.1136/jmg.40.5.305. Citation on PubMed or Free article on PubMed Central
- Wenger TL, Harr M, Ricciardi S, Bhoj E, Santani A, Adam MP, Barnett SS, Ganetzky R, McDonald-McGinn DM, Battaglia D, Bigoni S, Selicorni A, Sorge G, Monica MD, Mari F, Andreucci E, Romano S, Cocchi G, Savasta S, Malbora B, Marangi G, Garavelli L, Zollino M, Zackai EH. CHARGE-like presentation, craniosynostosis and mild Mowat-Wilson Syndrome diagnosed by recognition of the distinctive facial gestalt in a cohort of 28 new cases. Am J Med Genet A. 2014 Oct;164A(10):2557-66. doi: 10.1002/ajmg.a.36696. Epub 2014 Aug 14. Citation on PubMed
- Wilson M, Mowat D, Dastot-Le Moal F, Cacheux V, Kaariainen H, Cass D, Donnai D, Clayton-Smith J, Townshend S, Curry C, Gattas M, Braddock S, Kerr B, Aftimos S, Zehnwirth H, Barrey C, Goossens M. Further delineation of the phenotype associated with heterozygous mutations in ZFHX1B. Am J Med Genet A. 2003 Jun 15;119A(3):257-65. doi: 10.1002/ajmg.a.20053. Citation on PubMed
- Zollino M, Garavelli L, Rauch A. Clinical utility gene card for: Mowat-Wilson syndrome. Eur J Hum Genet. 2011 Aug;19(8). doi: 10.1038/ejhg.2011.12. Epub 2011 Feb 23. No abstract available. Citation on PubMed or Free article on PubMed Central
- Zweier C, Albrecht B, Mitulla B, Behrens R, Beese M, Gillessen-Kaesbach G, Rott HD, Rauch A. "Mowat-Wilson" syndrome with and without Hirschsprung disease is a distinct, recognizable multiple congenital anomalies-mental retardation syndrome caused by mutations in the zinc finger homeo box 1B gene. Am J Med Genet. 2002 Mar 15;108(3):177-81. Citation on PubMed
- Zweier C, Thiel CT, Dufke A, Crow YJ, Meinecke P, Suri M, Ala-Mello S, Beemer F, Bernasconi S, Bianchi P, Bier A, Devriendt K, Dimitrov B, Firth H, Gallagher RC, Garavelli L, Gillessen-Kaesbach G, Hudgins L, Kaariainen H, Karstens S, Krantz I, Mannhardt A, Medne L, Mucke J, Kibaek M, Krogh LN, Peippo M, Rittinger O, Schulz S, Schelley SL, Temple IK, Dennis NR, Van der Knaap MS, Wheeler P, Yerushalmi B, Zenker M, Seidel H, Lachmeijer A, Prescott T, Kraus C, Lowry RB, Rauch A. Clinical and mutational spectrum of Mowat-Wilson syndrome. Eur J Med Genet. 2005 Apr-Jun;48(2):97-111. doi: 10.1016/j.ejmg.2005.01.003. Epub 2005 Feb 25. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.