

Description
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) is a condition that affects many of the body's systems, particularly the brain and nervous system (encephalo-) and muscles (myopathy). The signs and symptoms of this disorder most often appear in childhood following a period of normal development, although they can begin at any age. Early symptoms may include muscle weakness and pain, recurrent headaches, loss of appetite, vomiting, and seizures. Most affected individuals experience stroke-like episodes beginning before age 40. These episodes often involve temporary muscle weakness on one side of the body (hemiparesis), altered consciousness, vision abnormalities, seizures, and severe headaches resembling migraines. Repeated stroke-like episodes can progressively damage the brain, leading to vision loss, problems with movement, and a loss of intellectual function (dementia).
Most people with MELAS have a buildup of lactic acid in their bodies, a condition called lactic acidosis. Increased acidity in the blood can lead to vomiting, abdominal pain, extreme tiredness (fatigue), muscle weakness, and difficulty breathing. Less commonly, people with MELAS may experience involuntary muscle spasms (myoclonus), impaired muscle coordination (ataxia), hearing loss, heart and kidney problems, diabetes, and hormonal imbalances.
Frequency
The exact incidence of MELAS is unknown. It is one of the more common conditions in a group known as mitochondrial diseases. Together, mitochondrial diseases occur in about 1 in 4,000 people.
Causes
MELAS can result from mutations in one of several genes, including MT-ND1, MT-ND5, MT-TH, MT-TL1, and MT-TV. These genes are found in the DNA of cellular structures called mitochondria, which convert the energy from food into a form that cells can use. Although most DNA is packaged in chromosomes within the nucleus, mitochondria also have a small amount of their own DNA, known as mitochondrial DNA or mtDNA.
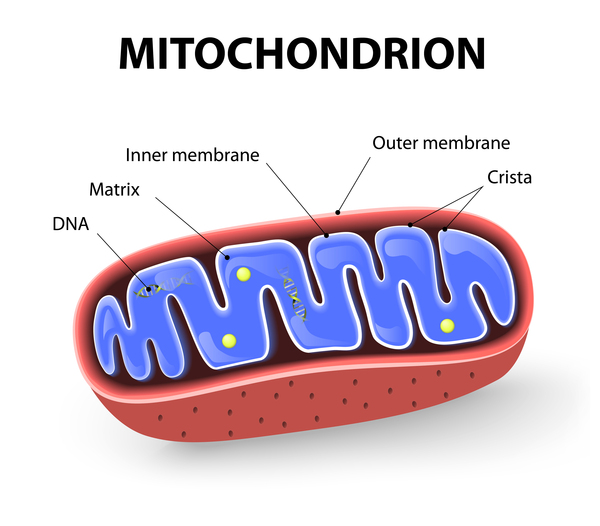
Some of the genes related to MELAS provide instructions for making proteins involved in normal mitochondrial function. These proteins are part of a large enzyme complex in mitochondria that helps convert oxygen, fats, and simple sugars to energy. Other genes associated with this disorder provide instructions for making molecules called transfer RNAs (tRNAs), which are chemical cousins of DNA. These molecules help assemble protein building blocks called amino acids into full-length, functioning proteins within mitochondria.



Mutations in a particular transfer RNA gene, MT-TL1, cause more than 80 percent of all cases of MELAS. These mutations impair the ability of mitochondria to make proteins, use oxygen, and produce energy. Researchers have not determined how changes in mtDNA lead to the specific signs and symptoms of MELAS. They continue to investigate the effects of mitochondrial gene mutations in different tissues, particularly in the brain.
Inheritance
This condition is inherited in a mitochondrial pattern, which is also known as maternal inheritance. This pattern of inheritance applies to genes contained in mtDNA. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can only inherit disorders resulting from mtDNA mutations from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits associated with changes in mtDNA to their children.
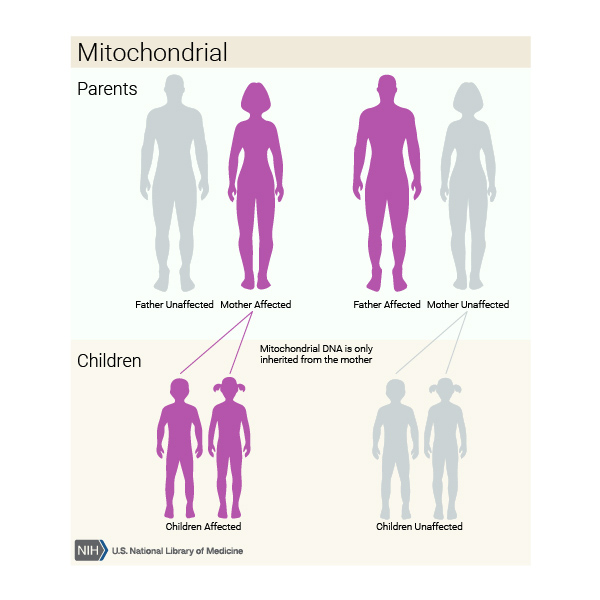
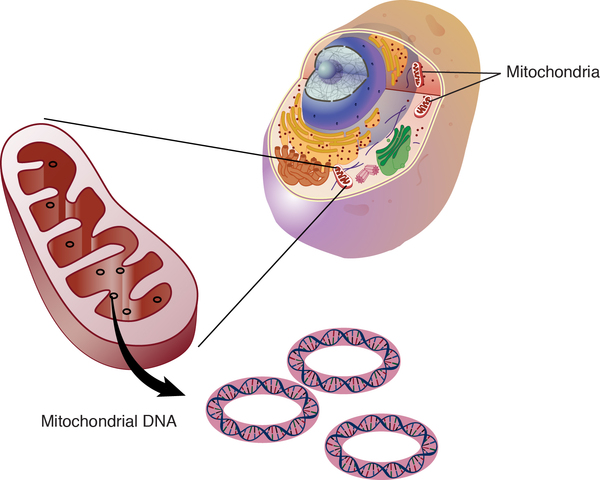
In most cases, people with MELAS inherit an altered mitochondrial gene from their mother. Less commonly, the disorder results from a new mutation in a mitochondrial gene and occurs in people with no family history of MELAS.
Other Names for This Condition
- MELAS
- MELAS syndrome
- Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes
- Mitochondrial myopathy, lactic acidosis, stroke-like episode
- Myopathy, mitochondrial-encephalopathy-lactic acidosis-stroke
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Betts J, Jaros E, Perry RH, Schaefer AM, Taylor RW, Abdel-All Z, Lightowlers RN, Turnbull DM. Molecular neuropathology of MELAS: level of heteroplasmy in individual neurones and evidence of extensive vascular involvement. Neuropathol Appl Neurobiol. 2006 Aug;32(4):359-73. doi: 10.1111/j.1365-2990.2006.00731.x. Citation on PubMed
- El-Hattab AW, Almannai M, Scaglia F. MELAS. 2001 Feb 27 [updated 2018 Nov 29]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1233/ Citation on PubMed
- Goodfellow JA, Dani K, Stewart W, Santosh C, McLean J, Mulhern S, Razvi S. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes: an important cause of stroke in young people. Postgrad Med J. 2012 Jun;88(1040):326-34. doi: 10.1136/postgradmedj-2011-130326. Epub 2012 Feb 10. Citation on PubMed
- Matsumoto J, Saver JL, Brennan KC, Ringman JM. Mitochondrial encephalomyopathy with lactic acidosis and stroke (MELAS). Rev Neurol Dis. 2005 Winter;2(1):30-4. Citation on PubMed
- Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann N Y Acad Sci. 2008 Oct;1142:133-58. doi: 10.1196/annals.1444.011. Citation on PubMed
- Thambisetty M, Newman NJ. Diagnosis and management of MELAS. Expert Rev Mol Diagn. 2004 Sep;4(5):631-44. doi: 10.1586/14737159.4.5.631. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.