

Description
Joubert syndrome is a condition that affects the development of the brain. This condition can also impact many other parts of the body. The signs and symptoms seen in people with Joubert syndrome can vary, even among members of the same family.
The hallmark feature of Joubert syndrome is a combination of brain abnormalities called the molar tooth sign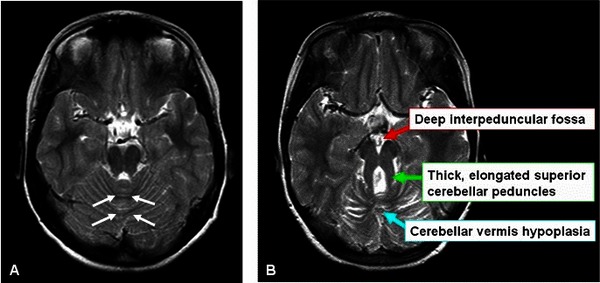 , which can be seen using magnetic resonance imaging (MRI). The molar tooth sign occurs when structures near the back of the brain, including the brainstem and the central part of the cerebellum (vermis), develop abnormally. This sign got its name because these brain abnormalities resemble the shape or outline of a molar tooth when seen on an MRI scan.
, which can be seen using magnetic resonance imaging (MRI). The molar tooth sign occurs when structures near the back of the brain, including the brainstem and the central part of the cerebellum (vermis), develop abnormally. This sign got its name because these brain abnormalities resemble the shape or outline of a molar tooth when seen on an MRI scan.
Infants with Joubert syndrome often have low muscle tone (hypotonia) and may experience breathing problems, with episodes of unusually fast (hyperpnea) or slow (apnea) breathing. Abnormal eye movements, which can include rapid, involuntary eye movements (nystagmus) or problems with side-to-side movements of the eyes (oculomotor apraxia), are also common.
Most individuals with Joubert syndrome have intellectual disabilities, which can range from mild to severe. Children with Joubert syndrome often have delayed development of speech and motor skills, such as sitting and walking. Problems with coordination and balance (ataxia) are common, but ataxia may become less severe over time. Most children with Joubert syndrome learn to walk on their own, although some affected individuals require walking support or wheelchair assistance.
Additional signs and symptoms that are seen in people with Joubert syndrome can include seizures, difficulty regulating body temperature, and behavioral problems. People with Joubert syndrome may have a broad range of other signs and symptoms. Kidney disease, liver disease, skeletal abnormalities (such as the presence of extra fingers and toes), or hormone (endocrine) problems may occur in affected individuals. Eye abnormalities may also be present and can include the breakdown of the light-sensitive tissue at the back of the eye (retinal dystrophy), which can cause vision loss.
In the past, individuals with the molar tooth sign were separated into different subgroups of Joubert syndrome based on their additional features. The term “Joubert syndrome and related disorders” (JSRD) was used to refer to these subgroups. Today, all people with the molar tooth sign, including those with additional signs and symptoms, are considered to have Joubert syndrome.
Frequency
Joubert syndrome is estimated to affect between 1 in 80,000 and 100,000 newborns. However, this estimate may be low because some affected individuals likely do not receive a diagnosis.
Causes
Joubert syndrome can be caused by variants (also called mutations) in one of more than 40 genes. The proteins produced from most of these genes are known or suspected to be involved with cell structures called primary cilia. Primary cilia are microscopic, finger-like projections that stick out from the surface of cells. They are part of signaling pathways that transmit information between cells. Primary cilia are important for the structure and function of many types of cells, including brain cells and certain cells in the kidneys and liver.
Variants in many of the genes that are associated with Joubert syndrome lead to problems with the structure and function of primary cilia, which can disrupt important signaling pathways during early development. Researchers believe that defective primary cilia are responsible for most of the features of Joubert syndrome.
Sixty-two to 94 percent of all people with Joubert syndrome have variants in one of the genes that are associated with this condition. In the remaining cases, the genetic cause is unknown.
Inheritance
Joubert syndrome can have different inheritance patterns depending on its genetic cause.
Joubert syndrome is almost always inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Joubert syndrome can also be inherited in an autosomal dominant pattern , which means one copy of the altered gene in each cell is sufficient to cause the disorder.
, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
In rare cases, the condition is inherited in an X-linked pattern. A condition is considered X-linked if the altered gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes in each cell. In males (who have only one X chromosome), a variant in the only copy of the gene in each cell is sufficient to cause the condition. In females (who have two copies of the X chromosome), one altered copy of the gene may or may not cause the condition. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
in each cell. In males (who have only one X chromosome), a variant in the only copy of the gene in each cell is sufficient to cause the condition. In females (who have two copies of the X chromosome), one altered copy of the gene may or may not cause the condition. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Other Names for This Condition
- Cerebellooculorenal disorder
- Cerebellooculorenal syndrome 1
- Cerebelloparenchymal disorder
- CORS
- CPD
- Familial aplasia of the vermis
- JBTS
- Joubert Syndrome and Related Disorders
- Joubert-Bolthauser syndrome
- JSRD
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- JOUBERT SYNDROME 1; JBTS1
- JOUBERT SYNDROME 10; JBTS10
- JOUBERT SYNDROME 2; JBTS2
- JOUBERT SYNDROME 3; JBTS3
- JOUBERT SYNDROME 5; JBTS5
- JOUBERT SYNDROME 6; JBTS6
- JOUBERT SYNDROME 9; JBTS9
- JOUBERT SYNDROME 8; JBTS8
- JOUBERT SYNDROME 4; JBTS4
- JOUBERT SYNDROME 7; JBTS7
- JOUBERT SYNDROME 23; JBTS23
- JOUBERT SYNDROME 36; JBTS36
- JOUBERT SYNDROME 20; JBTS20
- JOUBERT SYNDROME 17; JBTS17
- JOUBERT SYNDROME 21; JBTS21
- JOUBERT SYNDROME 24; JBTS24
- JOUBERT SYNDROME 15; JBTS15
- JOUBERT SYNDROME 16; JBTS16
- JOUBERT SYNDROME 27; JBTS27
- JOUBERT SYNDROME 28; JBTS28
- JOUBERT SYNDROME 13; JBTS13
- JOUBERT SYNDROME 35; JBTS35
- JOUBERT SYNDROME 25; JBTS25
- JOUBERT SYNDROME 26; JBTS26
- JOUBERT SYNDROME 22; JBTS22
- JOUBERT SYNDROME 18; JBTS18
- NEPHRONOPHTHISIS 14; NPHP14
- JOUBERT SYNDROME 30; JBTS30
- JOUBERT SYNDROME 32; JBTS32
- JOUBERT SYNDROME 31; JBTS31
- JOUBERT SYNDROME 33; JBTS33
- JOUBERT SYNDROME 14; JBTS14
- JOUBERT SYNDROME 37; JBTS37
- JOUBERT SYNDROME 38; JBTS38
- JOUBERT SYNDROME 40; JBTS40
- JOUBERT SYNDROME 39; JBTS39
Scientific Articles on PubMed
References
- Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis. 2010 Jul 8;5:20. doi: 10.1186/1750-1172-5-20. Citation on PubMed or Free article on PubMed Central
- Doherty D. Joubert syndrome: insights into brain development, cilium biology, and complex disease. Semin Pediatr Neurol. 2009 Sep;16(3):143-54. doi: 10.1016/j.spen.2009.06.002. Citation on PubMed or Free article on PubMed Central
- Gana S, Serpieri V, Valente EM. Genotype-phenotype correlates in Joubert syndrome: A review. Am J Med Genet C Semin Med Genet. 2022 Mar;190(1):72-88. doi: 10.1002/ajmg.c.31963. Epub 2022 Mar 3. Citation on PubMed
- Glass IA, Dempsey JC, Parisi M, Doherty D. Joubert Syndrome. 2003 Jul 9 [updated 2026 Feb 12]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1325/ Citation on PubMed
- Parisi MA. Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet C Semin Med Genet. 2009 Nov 15;151C(4):326-40. doi: 10.1002/ajmg.c.30229. Citation on PubMed or Free article on PubMed Central
- Pazour GJ. Cilia Structure and Function in Human Disease. Curr Opin Endocr Metab Res. 2024 Mar;34:100509. doi: 10.1016/j.coemr.2024.100509. Epub 2024 Feb 20. Citation on PubMed
- Valente EM, Dallapiccola B, Bertini E. Joubert syndrome and related disorders. Handb Clin Neurol. 2013;113:1879-88. doi: 10.1016/B978-0-444-59565-2.00058-7. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.