

Description
Horner syndrome is a disorder that affects the eye and surrounding tissues on one side of the face and results from paralysis of certain nerves. Horner syndrome can appear at any time of life; in about 5 percent of affected individuals, the disorder is present from birth (congenital).
Horner syndrome is characterized by drooping of the upper eyelid (ptosis) on the affected side, a constricted pupil in the affected eye (miosis) resulting in unequal pupil size (anisocoria), and absent sweating (anhidrosis) on the affected side of the face. Sinking of the eye into its cavity (enophthalmos) and a bloodshot eye often occur in this disorder. In people with Horner syndrome that occurs before the age of 2, the colored part (iris) of the eyes may differ in color (iris heterochromia), with the iris of the affected eye being lighter in color than that of the unaffected eye. Individuals who develop Horner syndrome after age 2 do not generally have iris heterochromia.
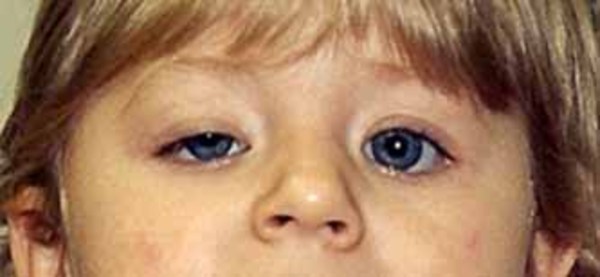
The abnormalities in the eye area related to Horner syndrome do not generally affect vision or health. However, the nerve damage that causes Horner syndrome may result from other health problems, some of which can be life-threatening.
Frequency
About 1 in 6,250 babies are born with Horner syndrome. The incidence of Horner syndrome that appears later is unknown, but it is considered an uncommon disorder.
Causes
Although congenital Horner syndrome can be passed down in families, no associated genes have been identified. Horner syndrome that appears after the newborn period (acquired Horner syndrome) and most cases of congenital Horner syndrome result from damage to nerves called the cervical sympathetics. These nerves belong to the part of the nervous system that controls involuntary functions (the autonomic nervous system). Within the autonomic nervous system, the nerves are part of a subdivision called the sympathetic nervous system. The cervical sympathetic nerves control several functions in the eye and face such as dilation of the pupil and sweating. Problems with the function of these nerves cause the signs and symptoms of Horner syndrome. Horner syndrome that occurs very early in life can lead to iris heterochromia because the development of the pigmentation (coloring) of the iris is under the control of the cervical sympathetic nerves.
Damage to the cervical sympathetic nerves can be caused by a direct injury to the nerves themselves, which can result from trauma that might occur during a difficult birth, surgery, or accidental injury. The nerves related to Horner syndrome can also be damaged by a benign or cancerous tumor, for example a childhood cancer of the nerve tissues called a neuroblastoma.
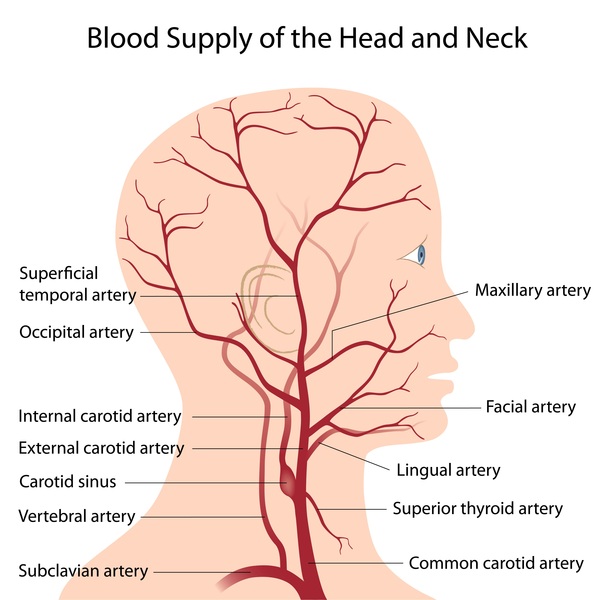
Horner syndrome can also be caused by problems with the artery that supplies blood to the head and neck (the carotid artery) on the affected side, resulting in loss of blood flow to the nerves. Some individuals with congenital Horner syndrome have a lack of development (agenesis) of the carotid artery. Tearing of the layers of the carotid artery wall (carotid artery dissection) can also lead to Horner syndrome.
The signs and symptoms of Horner syndrome can also occur during a migraine headache. When the headache is gone, the signs and symptoms of Horner syndrome usually also go away.
Some people with Horner syndrome have neither a known problem that would lead to nerve damage nor any history of the disorder in their family. These cases are referred to as idiopathic Horner syndrome.
Inheritance
Horner syndrome is usually not inherited and occurs in individuals with no history of the disorder in their family. Acquired Horner syndrome and most cases of congenital Horner syndrome have nongenetic causes. Rarely, congenital Horner syndrome is passed down within a family in a pattern that appears to be autosomal dominant, which means one copy of an altered gene in each cell is sufficient to cause the disorder. However, no genes associated with Horner syndrome have been identified.

Other Names for This Condition
- Bernard-Horner syndrome
- Horner's syndrome
- Oculosympathetic palsy
- Von Passow syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Fons C, Vasconcelos M, Vidal M, Puy R, Capdevila A, Sanchez L, Campistol J. Agenesis of internal carotid artery in a child with ipsilateral Horner's syndrome. J Child Neurol. 2009 Jan;24(1):101-4. doi: 10.1177/0883073808321049. Citation on PubMed
- Jeffery AR, Ellis FJ, Repka MX, Buncic JR. Pediatric Horner syndrome. J AAPOS. 1998 Jun;2(3):159-67. doi: 10.1016/s1091-8531(98)90008-8. Citation on PubMed
- Mahoney NR, Liu GT, Menacker SJ, Wilson MC, Hogarty MD, Maris JM. Pediatric horner syndrome: etiologies and roles of imaging and urine studies to detect neuroblastoma and other responsible mass lesions. Am J Ophthalmol. 2006 Oct;142(4):651-9. doi: 10.1016/j.ajo.2006.05.047. Citation on PubMed
- Renard D, Jeanjean L, Labauge P. Heterochromia Iridis in congenital Horner's syndrome. Eur Neurol. 2010;63(4):253. doi: 10.1159/000276946. Epub 2010 Apr 7. No abstract available. Citation on PubMed
- Smith SJ, Diehl N, Leavitt JA, Mohney BG. Incidence of pediatric Horner syndrome and the risk of neuroblastoma: a population-based study. Arch Ophthalmol. 2010 Mar;128(3):324-9. doi: 10.1001/archophthalmol.2010.6. Citation on PubMed or Free article on PubMed Central
- Walton KA, Buono LM. Horner syndrome. Curr Opin Ophthalmol. 2003 Dec;14(6):357-63. doi: 10.1097/00055735-200312000-00007. Citation on PubMed
- Wang FM, Wertenbaker C, Cho H, Marmor MA, Ahn-Lee SS, Bernard BA. Unilateral straight hair and congenital horner syndrome. J Neuroophthalmol. 2012 Jun;32(2):132-4. doi: 10.1097/WNO.0b013e318240c678. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.