

Description
Homocystinuria is an inherited disorder in which the body is unable to process certain building blocks of proteins (amino acids ) properly.
) properly.
The most common form of homocystinuria, called classic homocystinuria, is characterized by tall stature, nearsightedness (myopia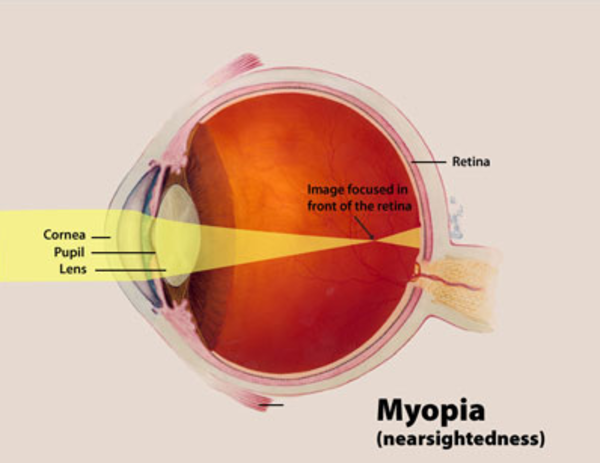 ), dislocation of the lens
), dislocation of the lens at the front of the eye, a higher risk of blood clotting disorders, and brittle bones that are prone to fracture (osteoporosis
at the front of the eye, a higher risk of blood clotting disorders, and brittle bones that are prone to fracture (osteoporosis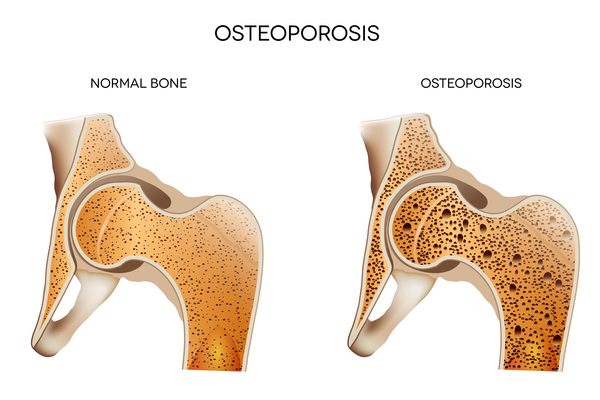 ) or other skeletal abnormalities. Some affected individuals also have developmental delay and learning problems.
) or other skeletal abnormalities. Some affected individuals also have developmental delay and learning problems.
Less common forms of homocystinuria can cause intellectual disability, slower growth and weight gain (failure to thrive), seizures, and problems with movement. They can also cause and a blood disorder called megaloblastic anemia,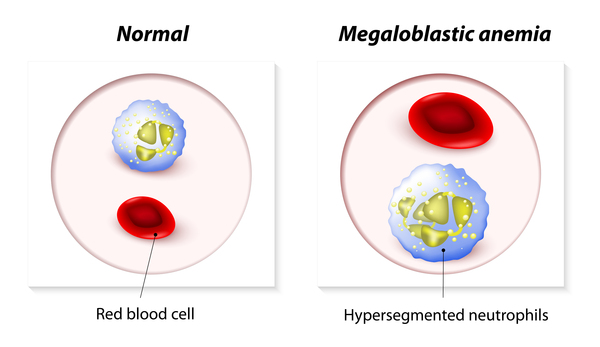 which occurs when a person has a low number of red blood cells (anemia), and the remaining red blood cells are larger than normal (megaloblastic).
which occurs when a person has a low number of red blood cells (anemia), and the remaining red blood cells are larger than normal (megaloblastic).
The signs and symptoms of homocystinuria typically develop during childhood, although some mildly affected people may not show signs and symptoms until adulthood.
Frequency
Classic homocystinuria affects at least 1 in 200,000 to 335,000 people worldwide. The disorder appears to be more common in some countries, such as Ireland (1 in 65,000), Germany (1 in 17,800), Norway (1 in 6,400), and Qatar (1 in 1,800). The rarer forms of homocystinuria each have a small number of cases reported in the scientific literature.
Causes
Variants (also called mutations) in the CBS, MTHFR, MTR, MTRR, and MMADHC genes cause homocystinuria.
Variants in the CBS gene cause classic homocystinuria. The CBS gene provides instructions for making an enzyme called cystathionine beta-synthase. This enzyme helps break down the amino acid methionine. Specifically, this enzyme is responsible for converting the amino acid homocysteine to a molecule called cystathionine. Variants in the CBS gene disrupt the function of cystathionine beta-synthase, preventing homocysteine from being used properly. As a result, homocysteine and methionine build up in the blood. Some of the excess homocysteine is excreted in urine.
Rarely, homocystinuria can be caused by variants in several other genes. The enzymes made by the MTHFR, MTR, MTRR, and MMADHC genes play roles in converting homocysteine to methionine. Variants in any of these genes prevent the enzymes from functioning properly, which leads to a buildup of homocysteine in the body. Researchers have not determined how excess homocysteine and related compounds lead to the signs and symptoms of homocystinuria.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Although people who carry one altered copy and one normal copy of the CBS gene do not have homocystinuria, they are more likely than people without a CBS variant to have shortages (deficiencies) of vitamin B12 and folic acid.
Other Names for This Condition
- Cystathionine beta synthase deficiency
- Homocysteinemia
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- HOMOCYSTINURIA DUE TO CYSTATHIONINE BETA-SYNTHASE DEFICIENCY
- HOMOCYSTINURIA DUE TO DEFICIENCY OF N(5,10)-METHYLENETETRAHYDROFOLATE REDUCTASE ACTIVITY
- HOMOCYSTINURIA-MEGALOBLASTIC ANEMIA, cblE TYPE; HMAE
- HOMOCYSTINURIA-MEGALOBLASTIC ANEMIA, cblG TYPE; HMAG
- METHYLMALONIC ACIDURIA AND HOMOCYSTINURIA, cblD TYPE; MAHCD
Scientific Articles on PubMed
References
- Adam S, Almeida MF, Carbasius Weber E, Champion H, Chan H, Daly A, Dixon M, Dokoupil K, Egli D, Evans S, Eyskens F, Faria A, Ferguson C, Hallam P, Heddrich-Ellerbrok M, Jacobs J, Jankowski C, Lachmann R, Lilje R, Link R, Lowry S, Luyten K, MacDonald A, Maritz C, Martins E, Meyer U, Muller E, Murphy E, Robertson LV, Rocha JC, Saruggia I, Schick P, Stafford J, Stoelen L, Terry A, Thom R, van den Hurk T, van Rijn M, van Teefelen-Heithoff A, Webster D, White FJ, Wildgoose J, Zweers H. Dietary practices in pyridoxine non-responsive homocystinuria: a European survey. Mol Genet Metab. 2013 Dec;110(4):454-9. doi: 10.1016/j.ymgme.2013.10.003. Epub 2013 Oct 10. Citation on PubMed
- Bublil EM, Majtan T. Classical homocystinuria: From cystathionine beta-synthase deficiency to novel enzyme therapies. Biochimie. 2020 Jun;173:48-56. doi: 10.1016/j.biochi.2019.12.007. Epub 2019 Dec 16. Citation on PubMed
- El-Said MF, Badii R, Bessisso MS, Shahbek N, El-Ali MG, El-Marikhie M, El-Zyoid M, Salem MS, Bener A, Hoffmann GF, Zschocke J. A common mutation in the CBS gene explains a high incidence of homocystinuria in the Qatari population. Hum Mutat. 2006 Jul;27(7):719. doi: 10.1002/humu.9436. Citation on PubMed
- Elsaid MF, Bener A, Lindner M, Alzyoud M, Shahbek N, Abdelrahman MO, Abdoh G, Bessisso MS, Zschocke J, Hoffmann GF. Are heterocygotes for classical homocystinuria at risk of vitamin B12 and folic acid deficiency? Mol Genet Metab. 2007 Sep-Oct;92(1-2):100-3. doi: 10.1016/j.ymgme.2007.06.010. Epub 2007 Aug 7. Citation on PubMed
- Kalantari S, Brezzi B, Bracciama V, Barreca A, Nozza P, Vaisitti T, Amoroso A, Deaglio S, Manganaro M, Porta F, Spada M. Adult-onset CblC deficiency: a challenging diagnosis involving different adult clinical specialists. Orphanet J Rare Dis. 2022 Feb 2;17(1):33. doi: 10.1186/s13023-022-02179-y. Citation on PubMed
- Majtan T, Kozich V, Kruger WD. Recent therapeutic approaches to cystathionine beta-synthase-deficient homocystinuria. Br J Pharmacol. 2023 Feb;180(3):264-278. doi: 10.1111/bph.15991. Epub 2022 Dec 8. Citation on PubMed
- Morrison T, Bosch F, Landolt MA, Kozich V, Huemer M, Morris AAM. Homocystinuria patient and caregiver survey: experiences of diagnosis and patient satisfaction. Orphanet J Rare Dis. 2021 Mar 10;16(1):124. doi: 10.1186/s13023-021-01764-x. Citation on PubMed
- Sacharow SJ, Levy HL. Homocystinuria due to Cystathionine Beta-Synthase Deficiency. 2004 Jan 15 [updated 2025 Sep 25]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1524/ Citation on PubMed
- Tran C, Bonafe L, Nuoffer JM, Rieger J, Berger MM. Adult classical homocystinuria requiring parenteral nutrition: Pitfalls and management. Clin Nutr. 2018 Aug;37(4):1114-1120. doi: 10.1016/j.clnu.2017.07.013. Epub 2017 Jul 25. Citation on PubMed
- Walter JH, Jahnke N, Remmington T. Newborn screening for homocystinuria. Cochrane Database Syst Rev. 2015 Oct 1;2015(10):CD008840. doi: 10.1002/14651858.CD008840.pub4. Citation on PubMed
- Watkins D, Rosenblatt DS. Inherited defects of cobalamin metabolism. Vitam Horm. 2022;119:355-376. doi: 10.1016/bs.vh.2022.01.010. Epub 2022 Feb 21. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.