

Description
Hartsfield syndrome is a rare condition characterized by holoprosencephaly, which is an abnormality of brain development, and a malformation of the hands and feet called ectrodactyly.
During early development before birth, the brain normally divides into two halves, the right and left hemispheres. Holoprosencephaly occurs when the brain fails to divide properly. In the most severe forms of holoprosencephaly, the brain does not divide at all. These affected individuals have one central eye (cyclopia) and a tubular nasal structure (proboscis) located above the eye. Most babies with severe holoprosencephaly die before birth or soon after. In less severe cases of holoprosencephaly, the brain is partially divided. The life expectancy of these affected individuals depends on the severity of signs and symptoms.
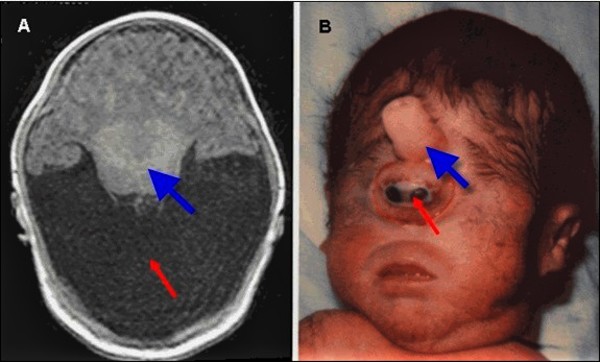
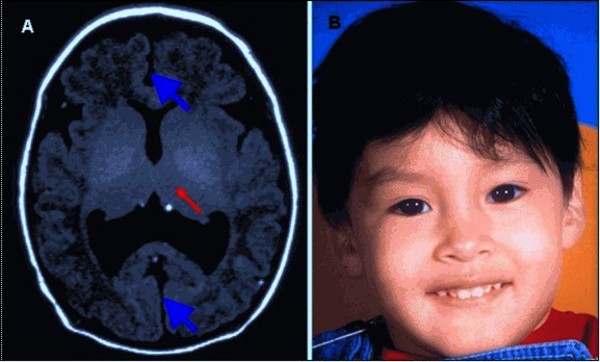
People with Hartsfield syndrome often have other brain abnormalities associated with holoprosencephaly. Affected individuals may have a malfunctioning pituitary, which is a gland located at the base of the brain that produces several hormones. Because pituitary dysfunction leads to the partial or complete absence of these hormones, it can cause a variety of disorders. These include diabetes insipidus, which disrupts the balance between fluid intake and urine excretion; a shortage (deficiency) of growth hormone, leading to slow or delayed growth; and hypogonadotropic hypogonadism, which affects the production of hormones that direct sexual development. Dysfunction in other parts of the brain can cause seizures, feeding difficulties, and problems regulating body temperature and sleep patterns. People with Hartsfield syndrome have delayed development that ranges from mild to severe.
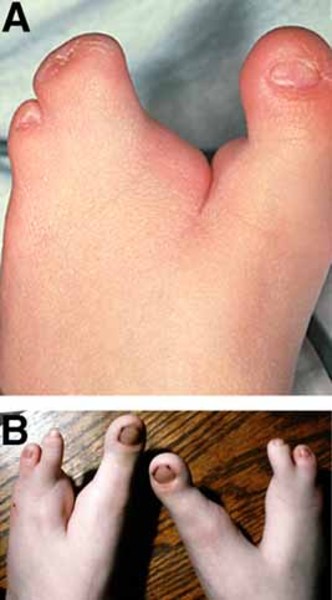
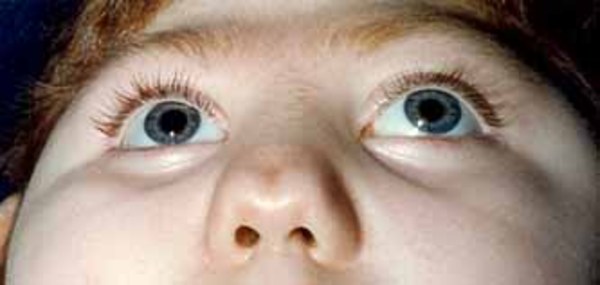
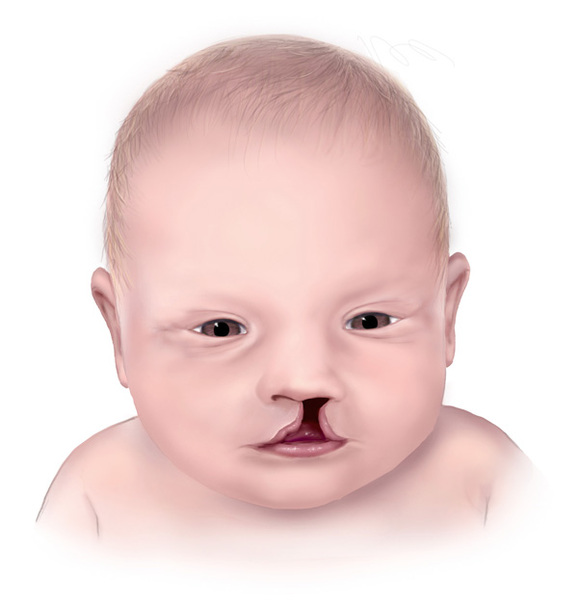
The other hallmark feature of Hartsfield syndrome is ectrodactyly. Ectrodactyly is a deep split in the hands, feet, or both, with missing fingers or toes and partial fusion of the remaining digits. It can affect the hands and feet on one or both sides. Other features that have been described in people with Hartsfield syndrome include premature fusion of certain bones of the skull (craniosynostosis), heart defects, abnormalities of the bones of the spine (vertebrae), and abnormal genitalia. Some affected individuals have distinctive facial features, including eyes that are widely spaced (hypertelorism) or closely spaced (hypotelorism), ears that are abnormally small or unusually shaped, and a split in the lip (cleft lip) with or without an opening in the roof of the mouth (cleft palate).



Frequency
Hartsfield syndrome appears to be a rare disorder. Fewer than 20 cases have been reported in the medical literature. For unknown reasons, most of the people who have been diagnosed with this disorder are male.
Causes
Hartsfield syndrome is caused by mutations in the FGFR1 gene, which provides instructions for making a protein called fibroblast growth factor receptor 1 (FGFR1). This receptor interacts with proteins called fibroblast growth factors (FGFs) to trigger signaling within cells. Signaling via the FGFR1 protein is involved in many critical processes, such as cell division and the regulation of cell growth and maturation. This signaling is important for the normal development and growth of several parts of the body, including the brain, bones of the head and face (craniofacial bones), and bones in the hands and feet.
The FGFR1 gene mutations that cause Hartsfield syndrome severely disrupt the function of the FGFR1 protein, including its ability to bind to FGFs. As a result, the receptor is unable to transmit signals properly, which impairs many aspects of normal development. It is unclear how these changes lead specifically to holoprosencephaly, ectrodactyly, and the other features of Hartsfield syndrome.
Some people with Hartsfield syndrome do not have an identified mutation in the FGFR1 gene. In these cases, the cause of the condition is unknown. Researchers are looking for other genetic changes that may also be associated with this disorder.
Inheritance
Hartsfield syndrome can have either an autosomal dominant or autosomal recessive pattern of inheritance. Autosomal dominant inheritance means one copy of the altered gene in each cell is sufficient to cause the disorder. In these cases, the condition usually results from a new (de novo) mutation in the FGFR1 gene that occurs during the formation of reproductive cells (eggs or sperm) or in early embryonic development. Most of these affected individuals have no history of the disorder in their family. However, in a small number of cases, people with Hartsfield syndrome have inherited the altered gene from an unaffected parent who has an FGFR1 gene mutation only in the sperm or egg cells. This phenomenon is called germline mosaicism.


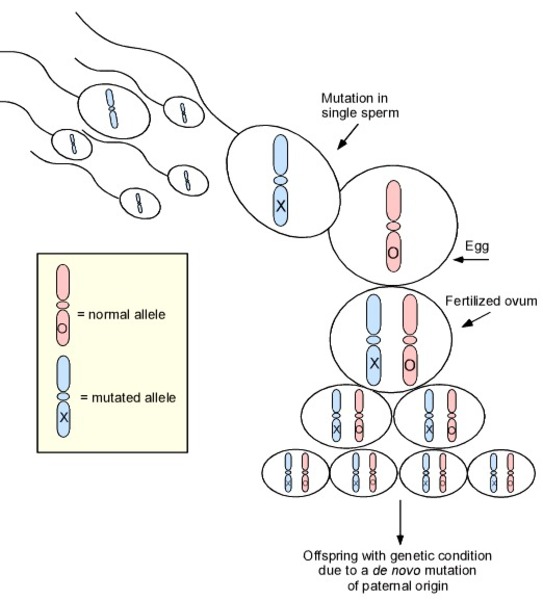
Less commonly, Hartsfield syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Hartsfield-Bixler-Demyer syndrome
- HHES
- Holoprosencephaly and split hand/foot syndrome
- Holoprosencephaly, ectrodactyly, and bilateral cleft lip/palate
- Holoprosencephaly, hypertelorism, and ectrodactyly syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Dhamija R, Babovic-Vuksanovic D. FGFR1-Related Hartsfield Syndrome. 2016 Mar 3 [updated 2022 May 12]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK349073/ Citation on PubMed
- Dhamija R, Kirmani S, Wang X, Ferber MJ, Wieben ED, Lazaridis KN, Babovic-Vuksanovic D. Novel de novo heterozygous FGFR1 mutation in two siblings with Hartsfield syndrome: a case of gonadal mosaicism. Am J Med Genet A. 2014 Sep;164A(9):2356-9. doi: 10.1002/ajmg.a.36621. Epub 2014 May 28. Citation on PubMed
- Hong S, Hu P, Marino J, Hufnagel SB, Hopkin RJ, Toromanovic A, Richieri-Costa A, Ribeiro-Bicudo LA, Kruszka P, Roessler E, Muenke M. Dominant-negative kinase domain mutations in FGFR1 can explain the clinical severity of Hartsfield syndrome. Hum Mol Genet. 2016 May 15;25(10):1912-1922. doi: 10.1093/hmg/ddw064. Epub 2016 Feb 29. Citation on PubMed
- Prasad R, Brewer C, Burren CP. Hartsfield syndrome associated with a novel heterozygous missense mutation in FGFR1 and incorporating tumoral calcinosis. Am J Med Genet A. 2016 Aug;170(8):2222-5. doi: 10.1002/ajmg.a.37731. Epub 2016 May 12. No abstract available. Citation on PubMed
- Shi Y, Dhamija R, Wren C, Wang X, Babovic-Vuksanovic D, Spector E. Detection of gonadal mosaicism in Hartsfield syndrome by next generation sequencing. Am J Med Genet A. 2016 Dec;170(12):3359. doi: 10.1002/ajmg.a.37869. Epub 2016 Sep 8. No abstract available. Citation on PubMed
- Simonis N, Migeotte I, Lambert N, Perazzolo C, de Silva DC, Dimitrov B, Heinrichs C, Janssens S, Kerr B, Mortier G, Van Vliet G, Lepage P, Casimir G, Abramowicz M, Smits G, Vilain C. FGFR1 mutations cause Hartsfield syndrome, the unique association of holoprosencephaly and ectrodactyly. J Med Genet. 2013 Sep;50(9):585-92. doi: 10.1136/jmedgenet-2013-101603. Epub 2013 Jun 28. Citation on PubMed or Free article on PubMed Central
- Vilain C, Mortier G, Van Vliet G, Dubourg C, Heinrichs C, de Silva D, Verloes A, Baumann C. Hartsfield holoprosencephaly-ectrodactyly syndrome in five male patients: further delineation and review. Am J Med Genet A. 2009 Jul;149A(7):1476-81. doi: 10.1002/ajmg.a.32678. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.