

Description
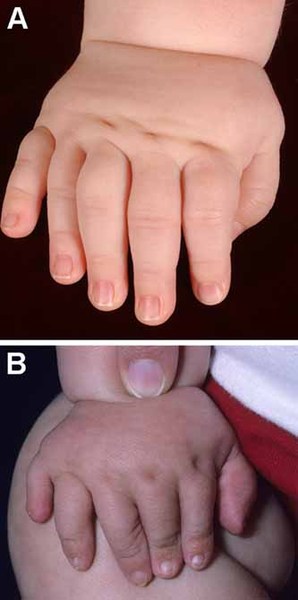
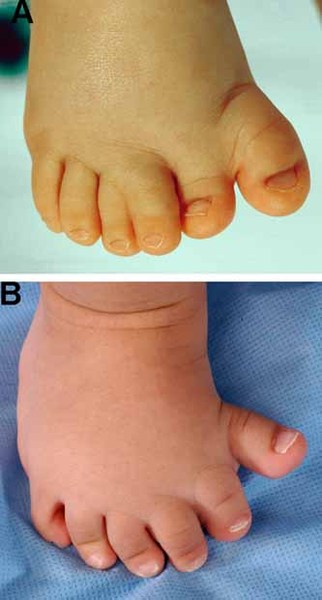
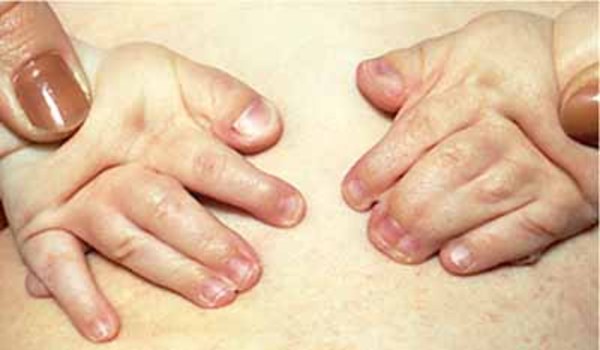
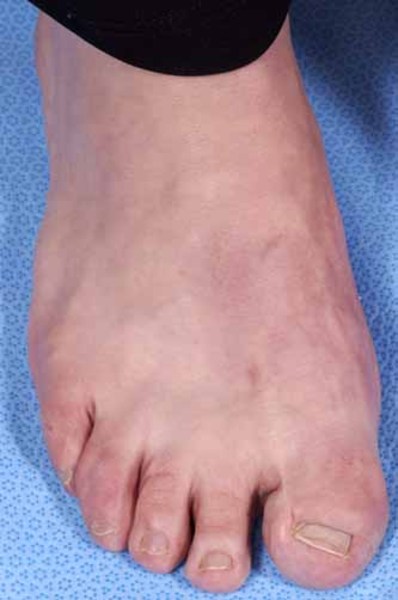
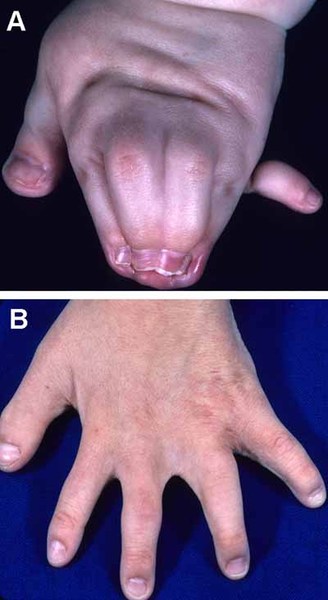
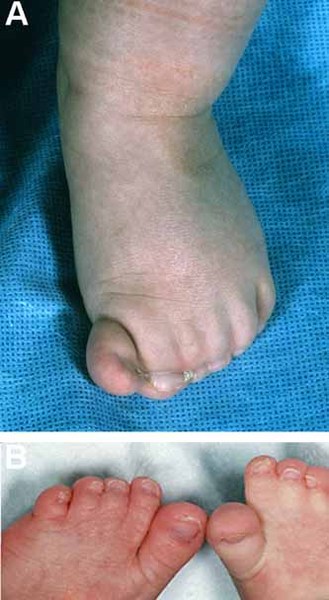
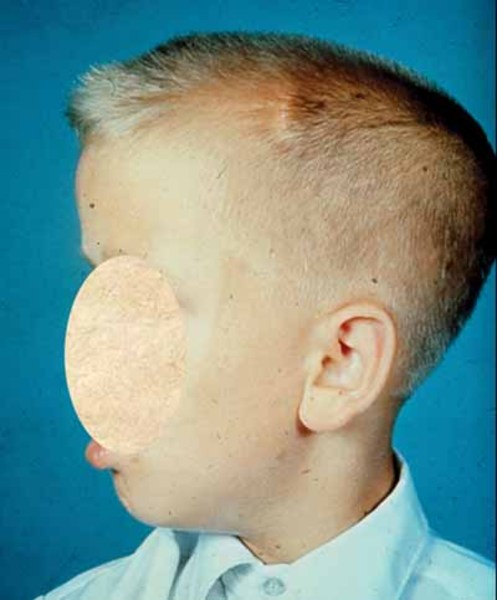
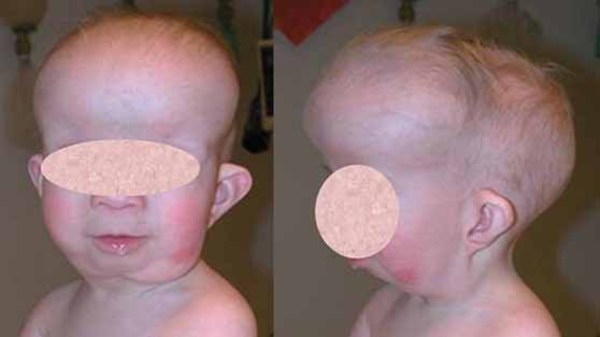
Greig cephalopolysyndactyly syndrome is a disorder that affects development of the limbs, head, and face. The features of this syndrome are highly variable, ranging from very mild to severe. People with this condition typically have one or more extra fingers or toes (polydactyly) or an abnormally wide thumb or big toe (hallux). The skin between the fingers and toes may be fused (cutaneous syndactyly). This disorder is also characterized by widely spaced eyes (ocular hypertelorism), an abnormally large head size (macrocephaly), and a high, prominent forehead. Rarely, affected individuals may have more serious medical problems including seizures, delayed development, and intellectual disability.

Frequency
This condition is very rare; its prevalence is unknown.
Causes
Mutations in the GLI3 gene cause Greig cephalopolysyndactyly syndrome. This gene provides instructions for making a protein that controls gene expression, which is a process that regulates whether genes are turned on or off in particular cells. By interacting with certain genes at specific times during development, the GLI3 protein plays a role in the normal shaping (patterning) of many organs and tissues before birth.
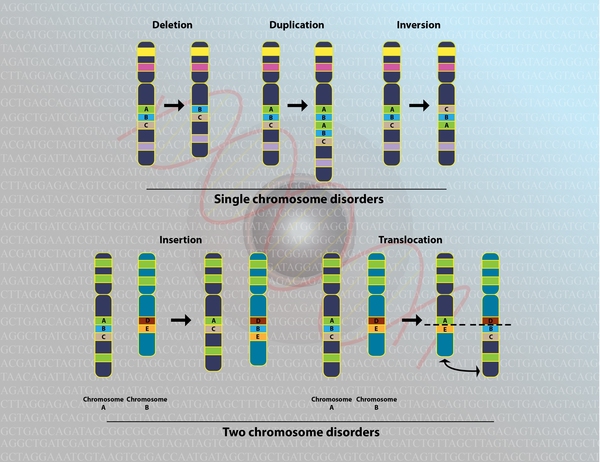
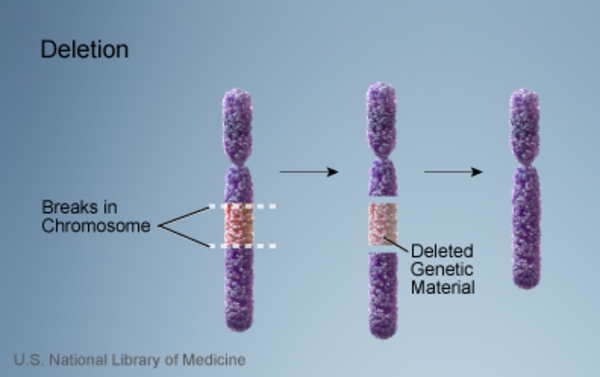
Different genetic changes involving the GLI3 gene can cause Greig cephalopolysyndactyly syndrome. In some cases, the condition results from a chromosomal abnormality—such as a large deletion or rearrangement of genetic material—in the region of chromosome 7 that contains the GLI3 gene. In other cases, a mutation in the GLI3 gene itself is responsible for the disorder. Each of these genetic changes prevents one copy of the gene in each cell from producing any functional protein. It is unclear how a reduced amount of this protein disrupts early development and causes the characteristic features of Greig cephalopolysyndactyly syndrome.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one altered or missing copy of the GLI3 gene in each cell is sufficient to cause the disorder. In some cases, an affected person inherits a gene mutation or chromosomal abnormality from one affected parent. Other cases occur in people with no history of the condition in their family.


Other Names for This Condition
- Cephalopolysyndactyly syndrome
- GCPS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Biesecker LG, Johnston JJ. GLI3-Related Greig Cephalopolysyndactyly Syndrome. 2001 Jul 9 [updated 2025 Aug 28]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1446/ Citation on PubMed
- Biesecker LG. The Greig cephalopolysyndactyly syndrome. Orphanet J Rare Dis. 2008 Apr 24;3:10. doi: 10.1186/1750-1172-3-10. Citation on PubMed or Free article on PubMed Central
- Debeer P, Peeters H, Driess S, De Smet L, Freese K, Matthijs G, Bornholdt D, Devriendt K, Grzeschik KH, Fryns JP, Kalff-Suske M. Variable phenotype in Greig cephalopolysyndactyly syndrome: clinical and radiological findings in 4 independent families and 3 sporadic cases with identified GLI3 mutations. Am J Med Genet A. 2003 Jul 1;120A(1):49-58. doi: 10.1002/ajmg.a.20018. Citation on PubMed
- Johnston JJ, Olivos-Glander I, Killoran C, Elson E, Turner JT, Peters KF, Abbott MH, Aughton DJ, Aylsworth AS, Bamshad MJ, Booth C, Curry CJ, David A, Dinulos MB, Flannery DB, Fox MA, Graham JM, Grange DK, Guttmacher AE, Hannibal MC, Henn W, Hennekam RC, Holmes LB, Hoyme HE, Leppig KA, Lin AE, Macleod P, Manchester DK, Marcelis C, Mazzanti L, McCann E, McDonald MT, Mendelsohn NJ, Moeschler JB, Moghaddam B, Neri G, Newbury-Ecob R, Pagon RA, Phillips JA, Sadler LS, Stoler JM, Tilstra D, Walsh Vockley CM, Zackai EH, Zadeh TM, Brueton L, Black GC, Biesecker LG. Molecular and clinical analyses of Greig cephalopolysyndactyly and Pallister-Hall syndromes: robust phenotype prediction from the type and position of GLI3 mutations. Am J Hum Genet. 2005 Apr;76(4):609-22. doi: 10.1086/429346. Epub 2005 Feb 28. Citation on PubMed or Free article on PubMed Central
- Johnston JJ, Olivos-Glander I, Turner J, Aleck K, Bird LM, Mehta L, Schimke RN, Heilstedt H, Spence JE, Blancato J, Biesecker LG. Clinical and molecular delineation of the Greig cephalopolysyndactyly contiguous gene deletion syndrome and its distinction from acrocallosal syndrome. Am J Med Genet A. 2003 Dec 15;123A(3):236-42. doi: 10.1002/ajmg.a.20318. Citation on PubMed
- Johnston JJ, Sapp JC, Turner JT, Amor D, Aftimos S, Aleck KA, Bocian M, Bodurtha JN, Cox GF, Curry CJ, Day R, Donnai D, Field M, Fujiwara I, Gabbett M, Gal M, Graham JM, Hedera P, Hennekam RC, Hersh JH, Hopkin RJ, Kayserili H, Kidd AM, Kimonis V, Lin AE, Lynch SA, Maisenbacher M, Mansour S, McGaughran J, Mehta L, Murphy H, Raygada M, Robin NH, Rope AF, Rosenbaum KN, Schaefer GB, Shealy A, Smith W, Soller M, Sommer A, Stalker HJ, Steiner B, Stephan MJ, Tilstra D, Tomkins S, Trapane P, Tsai AC, Van Allen MI, Vasudevan PC, Zabel B, Zunich J, Black GC, Biesecker LG. Molecular analysis expands the spectrum of phenotypes associated with GLI3 mutations. Hum Mutat. 2010 Oct;31(10):1142-54. doi: 10.1002/humu.21328. Citation on PubMed or Free article on PubMed Central
- Kalff-Suske M, Wild A, Topp J, Wessling M, Jacobsen EM, Bornholdt D, Engel H, Heuer H, Aalfs CM, Ausems MG, Barone R, Herzog A, Heutink P, Homfray T, Gillessen-Kaesbach G, Konig R, Kunze J, Meinecke P, Muller D, Rizzo R, Strenge S, Superti-Furga A, Grzeschik KH. Point mutations throughout the GLI3 gene cause Greig cephalopolysyndactyly syndrome. Hum Mol Genet. 1999 Sep;8(9):1769-77. doi: 10.1093/hmg/8.9.1769. Citation on PubMed
- Kroisel PM, Petek E, Wagner K. Phenotype of five patients with Greig syndrome and microdeletion of 7p13. Am J Med Genet. 2001 Aug 15;102(3):243-9. doi: 10.1002/ajmg.1443. Citation on PubMed
- Wild A, Kalff-Suske M, Vortkamp A, Bornholdt D, Konig R, Grzeschik KH. Point mutations in human GLI3 cause Greig syndrome. Hum Mol Genet. 1997 Oct;6(11):1979-84. doi: 10.1093/hmg/6.11.1979. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.