

Description
Gorlin syndrome, also known as nevoid basal cell carcinoma syndrome, is a condition that affects many areas of the body and increases the risk of developing various cancerous and noncancerous tumors.
In people with Gorlin syndrome, the type of cancer diagnosed most often is basal cell carcinoma, which is the most common form of skin cancer. Individuals with Gorlin syndrome typically begin to develop basal cell carcinomas during adolescence or early adulthood. These cancers occur most often on the face, chest, and back. The number of basal cell carcinomas that develop during a person's lifetime varies among affected individuals. Some people with Gorlin syndrome never develop any basal cell carcinomas, while others may develop thousands of these cancers. Individuals with lighter skin are more likely to develop basal cell carcinomas than are people with darker skin. The number of carcinomas may be reduced with ongoing treatment.
Most people with Gorlin syndrome also develop noncancerous (benign) tumors of the jaw, called keratocystic odontogenic tumors. These tumors usually first appear during adolescence, and new tumors form until about age 30. Keratocystic odontogenic tumors rarely develop later in adulthood. If untreated, these tumors may cause painful facial swelling and tooth displacement.
Individuals with Gorlin syndrome have a higher risk than the general population of developing other tumors. A small proportion of affected individuals develop a brain tumor called medulloblastoma during childhood. A type of benign tumor called a fibroma can occur in the heart or in a woman's ovaries. Heart (cardiac) fibromas often do not cause any symptoms, but they may obstruct blood flow or cause irregular heartbeats (arrhythmia). Ovarian fibromas are not thought to affect a woman's ability to have children (fertility).
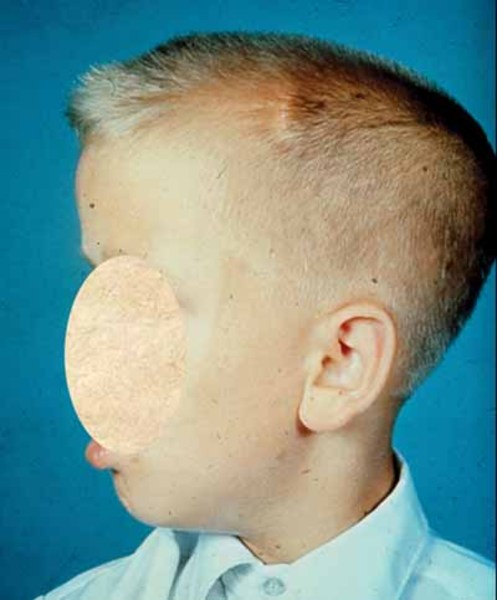
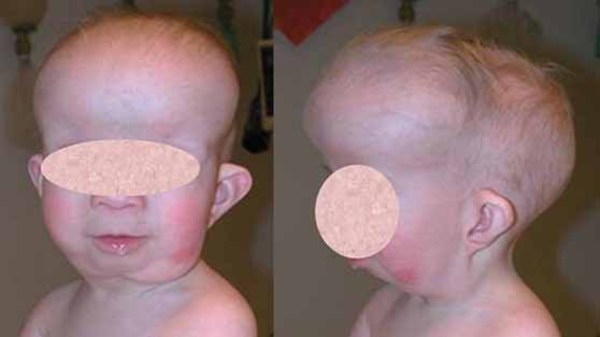
Other features of Gorlin syndrome include small depressions (pits) in the skin of the palms of the hands and soles of the feet; an unusually large head size (macrocephaly) with a prominent forehead; and skeletal abnormalities involving the spine, ribs, or skull. These signs and symptoms are typically apparent from birth or become evident in early childhood.
Frequency
Gorlin syndrome affects an estimated 1 in 31,000 people. While more than 1 million new cases of basal cell carcinoma are diagnosed each year in the United States, fewer than 1 percent of these skin cancers are related to Gorlin syndrome.
Causes
Mutations in the PTCH1 gene are the main cause Gorlin syndrome. This gene provides instructions for making a protein called patched-1, which functions as a receptor. Receptor proteins have specific sites into which certain other proteins, called ligands, fit like keys into locks. Together, ligands and their receptors trigger signals that affect cell development and function. A protein called Sonic Hedgehog is the ligand for the patched-1 receptor. Patched-1 blocks cell growth and division (proliferation) until Sonic Hedgehog is attached.
The PTCH1 gene is a tumor suppressor gene, which means it stops cells from proliferating too rapidly or in an uncontrolled way. Mutations in this gene prevent the production of patched-1 or lead to the production of an abnormal version of the receptor. An altered or missing patched-1 receptor cannot effectively suppress cell growth and division. As a result, cells proliferate uncontrollably to form the tumors that are characteristic of Gorlin syndrome. It is less clear how PTCH1 gene mutations cause the other signs and symptoms related to this condition.
The characteristic features of Gorlin syndrome can also be associated with a chromosomal change called a 9q22.3 microdeletion, in which a small piece of chromosome 9 is deleted in each cell. This deletion includes the segment of chromosome 9 that contains the PTCH1 gene, and as a result, people with a 9q22.3 microdeletion are missing one copy of this gene. Loss of this gene underlies the signs and symptoms of Gorlin syndrome in people with 9q22.3 microdeletions. Affected individuals also have features that are not typically associated with Gorlin syndrome, including delayed development, intellectual disability, overgrowth of the body (macrosomia), and other physical abnormalities. Researchers believe that these other signs and symptoms may result from the loss of additional genes in the deleted region of chromosome 9.
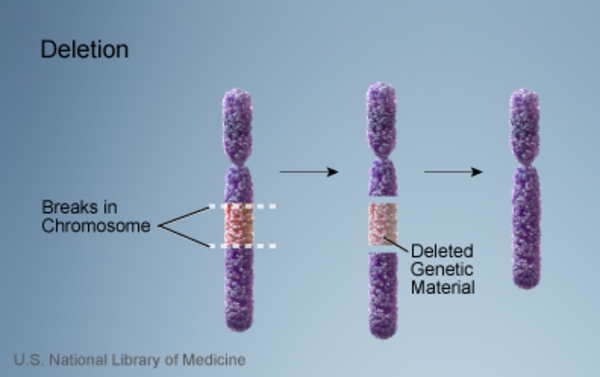
Mutations in the PTCH1 gene account for 50 to 85 percent of cases of Gorlin syndrome, while mutations in other genes, some known and some unidentified, are responsible for the remaining cases.
Inheritance
Gorlin syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the condition. In most cases, an affected person inherits the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.


Having one mutated copy of the PTCH1 gene in each cell is enough to cause the features of Gorlin syndrome that are present early in life, including macrocephaly and skeletal abnormalities. For basal cell carcinomas and other tumors to develop, a mutation in the second copy of the PTCH1 gene must also occur in certain cells during the person's lifetime. Most people who are born with one PTCH1 gene mutation eventually acquire a second mutation in some cells and consequently develop various types of tumors.
Other Names for This Condition
- Basal cell nevus syndrome
- BCNS
- Gorlin-Goltz syndrome
- NBCCS
- Nevoid basal cell carcinoma syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bale AE, Yu KP. The hedgehog pathway and basal cell carcinomas. Hum Mol Genet. 2001 Apr;10(7):757-62. doi: 10.1093/hmg/10.7.757. Citation on PubMed
- Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, Lalloo F. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A. 2010 Feb;152A(2):327-32. doi: 10.1002/ajmg.a.33139. Citation on PubMed
- Evans DG. Nevoid Basal Cell Carcinoma Syndrome. 2002 Jun 20 [updated 2024 Feb 22]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1151/ Citation on PubMed
- Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med. 2004 Nov-Dec;6(6):530-9. doi: 10.1097/01.gim.0000144188.15902.c4. No abstract available. Citation on PubMed
- Guenther LC, Barber K, Searles GE, Lynde CW, Janiszewski P, Ashkenas J; Canadian Non-melanoma Skin Cancer Guidelines Committee. Non-melanoma Skin Cancer in Canada Chapter 1: Introduction to the Guidelines. J Cutan Med Surg. 2015 May-Jun;19(3):205-15. doi: 10.1177/1203475415588652. Epub 2015 May 27. Citation on PubMed
- High A, Zedan W. Basal cell nevus syndrome. Curr Opin Oncol. 2005 Mar;17(2):160-6. doi: 10.1097/01.cco.0000154108.99236.ed. Citation on PubMed
- Lo Muzio L, Pastorino L, Levanat S, Musani V, Situm M, Scarra GB. Clinical utility gene card for: Gorlin syndrome. Eur J Hum Genet. 2011 Aug;19(8). doi: 10.1038/ejhg.2011.9. Epub 2011 Feb 9. No abstract available. Citation on PubMed or Free article on PubMed Central
- Madras J, Lapointe H. Keratocystic odontogenic tumour: reclassification of the odontogenic keratocyst from cyst to tumour. J Can Dent Assoc. 2008 Mar;74(2):165-165h. Citation on PubMed
- Muller EA, Aradhya S, Atkin JF, Carmany EP, Elliott AM, Chudley AE, Clark RD, Everman DB, Garner S, Hall BD, Herman GE, Kivuva E, Ramanathan S, Stevenson DA, Stockton DW, Hudgins L. Microdeletion 9q22.3 syndrome includes metopic craniosynostosis, hydrocephalus, macrosomia, and developmental delay. Am J Med Genet A. 2012 Feb;158A(2):391-9. doi: 10.1002/ajmg.a.34216. Epub 2011 Dec 21. Citation on PubMed
- Smith MJ, Beetz C, Williams SG, Bhaskar SS, O'Sullivan J, Anderson B, Daly SB, Urquhart JE, Bholah Z, Oudit D, Cheesman E, Kelsey A, McCabe MG, Newman WG, Evans DG. Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol. 2014 Dec 20;32(36):4155-61. doi: 10.1200/JCO.2014.58.2569. Epub 2014 Nov 17. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.