

Description
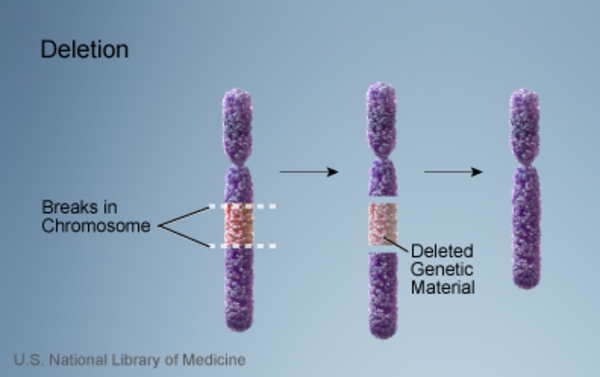
9q22.3 microdeletion is a chromosomal change in which a small piece of chromosome 9 is deleted in each cell. The deletion occurs on the long (q) arm of the chromosome in a region designated q22.3. This chromosomal change is associated with delayed development, intellectual disability, certain physical abnormalities, and the characteristic features of a genetic condition called Gorlin syndrome.
Many individuals with a 9q22.3 microdeletion have delayed development, particularly affecting the development of motor skills such as sitting, standing, and walking. In some people, the delays are temporary and improve in childhood. More severely affected individuals have permanent developmental disabilities along with intellectual impairment and learning problems. Rarely, seizures have been reported in people with a 9q22.3 microdeletion.
About 20 percent of people with a 9q22.3 microdeletion experience overgrowth (macrosomia), which results in increased height and weight compared to unaffected peers. The macrosomia often begins before birth and continues into childhood. Other physical changes that are sometimes associated with a 9q22.3 microdeletion include the premature fusion of certain bones in the skull (metopic craniosynostosis) and a buildup of fluid in the brain (hydrocephalus). Affected individuals can also have distinctive facial features such as a prominent forehead with vertical skin creases, upward- or downward-slanting eyes, a short nose, and a long space between the nose and upper lip (philtrum).
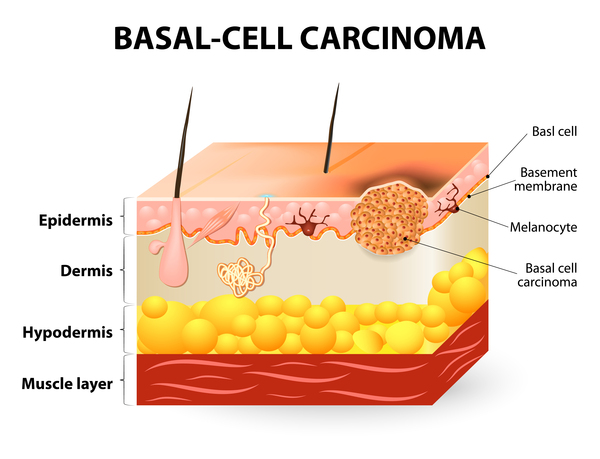
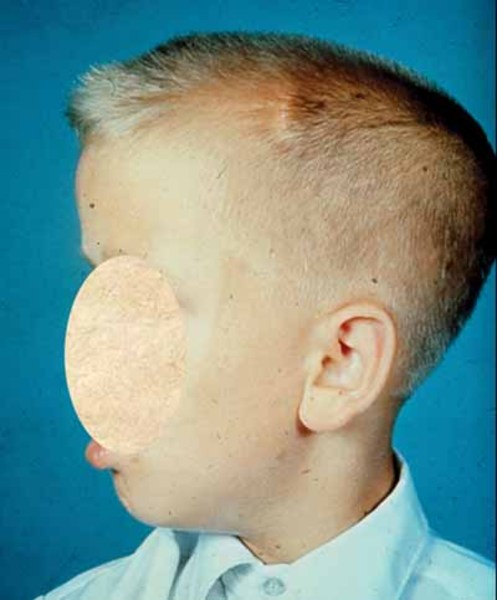
9q22.3 microdeletions also cause the characteristic features of Gorlin syndrome (also known as nevoid basal cell carcinoma syndrome). This genetic condition affects many areas of the body and increases the risk of developing various cancerous and noncancerous tumors. In people with Gorlin syndrome, the type of cancer diagnosed most often is basal cell carcinoma, which is the most common form of skin cancer. Most people with this condition also develop noncancerous (benign) tumors of the jaw, called keratocystic odontogenic tumors, which can cause facial swelling and tooth displacement. Other types of tumors that occur in some people with Gorlin syndrome include a form of childhood brain cancer called a medulloblastoma and a type of benign tumor called a fibroma that occurs in the heart or in a woman's ovaries. Other features of Gorlin syndrome include small depressions (pits) in the skin of the palms of the hands and soles of the feet; an unusually large head size (macrocephaly) with a prominent forehead; and skeletal abnormalities involving the spine, ribs, or skull.
Frequency
9q22.3 microdeletion appears to be a rare chromosomal change. About three dozen affected individuals have been reported in the medical literature.
Causes
People with a 9q22.3 microdeletion are missing a sequence of at least 352,000 DNA building blocks (base pairs), also written as 352 kilobases (kb), in the q22.3 region of chromosome 9. This 352-kb segment is known as the minimum critical region because it is the smallest deletion that has been found to cause the signs and symptoms described above. 9q22.3 microdeletions can also be much larger; the largest reported deletion includes 20.5 million base pairs (20.5 Mb). 9q22.3 microdeletion affects one of the two copies of chromosome 9 in each cell.
People with a 9q22.3 microdeletion are missing from two to more than 270 genes on chromosome 9. All known 9q22.3 microdeletions include the PTCH1 gene. The protein produced from this gene, patched-1, acts as a tumor suppressor, which means it keeps cells from growing and dividing (proliferating) too rapidly or in an uncontrolled way. Researchers believe that many of the features associated with 9q22.3 microdeletions, particularly the signs and symptoms of Gorlin syndrome, result from a loss of the PTCH1 gene. When this gene is missing, patched-1 is not available to suppress cell proliferation. As a result, cells divide uncontrollably to form the tumors that are characteristic of Gorlin syndrome.
Other signs and symptoms related to 9q22.3 microdeletions probably result from the loss of additional genes in the q22.3 region. Researchers are working to determine which missing genes contribute to the other features associated with the deletion.
Inheritance
9q22.3 microdeletions are inherited in an autosomal dominant pattern, which means that missing genetic material from one of the two copies of chromosome 9 in each cell is sufficient to cause delayed development, intellectual disability, and the features of Gorlin syndrome.
A 9q22.3 microdeletion most often occurs in people whose parents do not carry the chromosomal change. In these cases, the deletion occurs as a random (de novo) event during the formation of reproductive cells (eggs or sperm) in a parent or in early embryonic development. De novo chromosomal changes occur in people with no history of the disorder in their family.

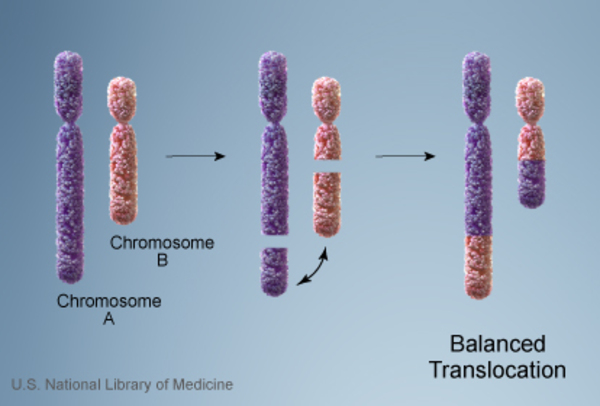
Less commonly, individuals with a 9q22.3 microdeletion inherit the chromosomal change from an unaffected parent. In these cases, the parent carries a chromosomal rearrangement called a balanced translocation, in which a segment of chromosome 9 has traded places with a segment of another chromosome. No genetic material is gained or lost in a balanced translocation, so these chromosomal changes usually do not cause any health problems. However, translocations can become unbalanced as they are passed to the next generation. People who inherit a 9q22.3 microdeletion receive an unbalanced translocation that deletes genetic material from one copy of the q22.3 region of chromosome 9 in each cell.
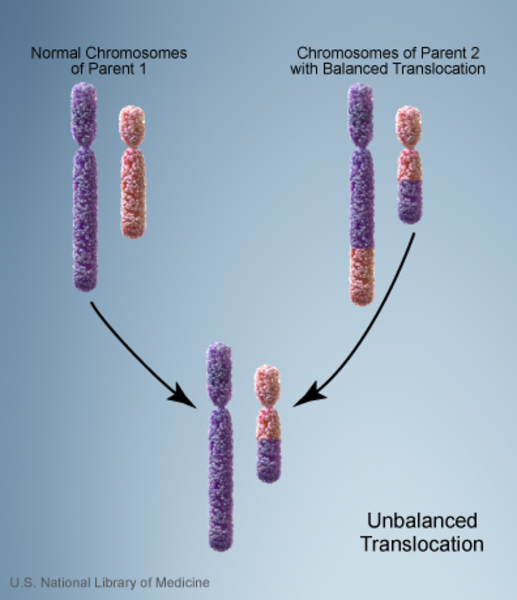
Having one missing copy of the PTCH1 gene in each cell is enough to cause the features of Gorlin syndrome that are present early in life, including macrocephaly and skeletal abnormalities. For basal cell carcinomas and other tumors to develop, a mutation in the other copy of the PTCH1 gene must also occur in certain cells during the person's lifetime. Most people who are born with one missing copy of the PTCH1 gene eventually acquire a mutation in the other copy of the gene in some cells and consequently develop various types of tumors.
Other Names for This Condition
- 9q22 deletion syndrome
- 9q22.3 deletion
- Microdeletion 9q22.3 syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Evans DG. Nevoid Basal Cell Carcinoma Syndrome. 2002 Jun 20 [updated 2024 Feb 22]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1151/ Citation on PubMed
- Muller EA, Aradhya S, Atkin JF, Carmany EP, Elliott AM, Chudley AE, Clark RD, Everman DB, Garner S, Hall BD, Herman GE, Kivuva E, Ramanathan S, Stevenson DA, Stockton DW, Hudgins L. Microdeletion 9q22.3 syndrome includes metopic craniosynostosis, hydrocephalus, macrosomia, and developmental delay. Am J Med Genet A. 2012 Feb;158A(2):391-9. doi: 10.1002/ajmg.a.34216. Epub 2011 Dec 21. Citation on PubMed
- Redon R, Baujat G, Sanlaville D, Le Merrer M, Vekemans M, Munnich A, Carter NP, Cormier-Daire V, Colleaux L. Interstitial 9q22.3 microdeletion: clinical and molecular characterisation of a newly recognised overgrowth syndrome. Eur J Hum Genet. 2006 Jun;14(6):759-67. doi: 10.1038/sj.ejhg.5201613. Citation on PubMed
- Shimojima K, Adachi M, Tanaka M, Tanaka Y, Kurosawa K, Yamamoto T. Clinical features of microdeletion 9q22.3 (pat). Clin Genet. 2009 Apr;75(4):384-93. doi: 10.1111/j.1399-0004.2008.01141.x. Citation on PubMed
- Yamamoto K, Yoshihashi H, Furuya N, Adachi M, Ito S, Tanaka Y, Masuno M, Chiyo H, Kurosawa K. Further delineation of 9q22 deletion syndrome associated with basal cell nevus (Gorlin) syndrome: report of two cases and review of the literature. Congenit Anom (Kyoto). 2009 Mar;49(1):8-14. doi: 10.1111/j.1741-4520.2008.00212.x. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.