

Description
Gorlin-Chaudhry-Moss syndrome is a condition that affects many parts of the body. The signs and symptoms of this disorder are apparent from birth or infancy.
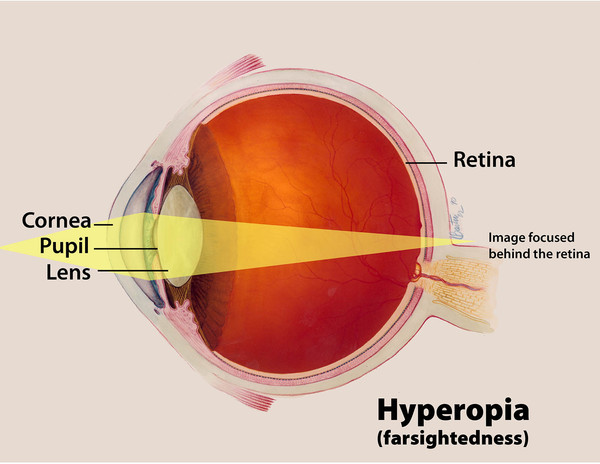
Gorlin-Chaudhry-Moss syndrome is characterized by the premature closure of certain bones of the skull (craniosynostosis) during development, which affects the shape of the head and face. Many people with this disorder have a premature fusion of skull bones along the coronal suture, the growth line that goes over the head from ear to ear. These changes can result in a head that is abnormally wide and pointed at the top (acrobrachycephaly). Affected individuals also have distinctive facial characteristics that can include a flat or sunken appearance of the middle of the face (midface hypoplasia), and small eyes (microphthalmia) with narrowed openings (narrowed palpebral fissures). Affected individuals may also have farsightedness (hyperopia) and dental problems such as small teeth (microdontia) or fewer teeth than normal (hypodontia).

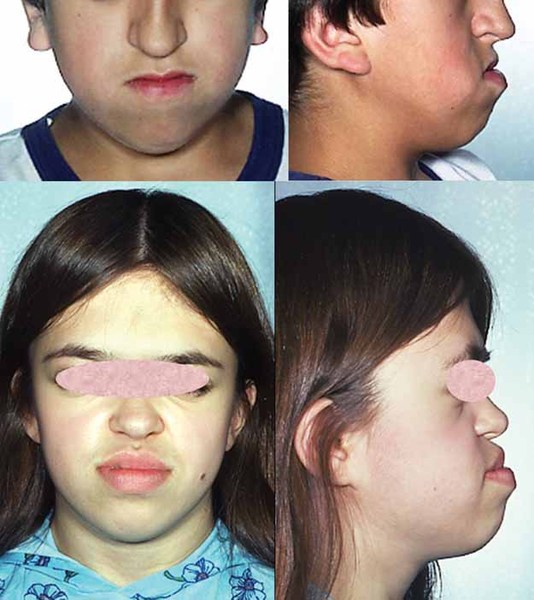
Many people with Gorlin-Chaudhry-Moss syndrome have a lack of fatty tissue under the skin (lipodystrophy). The lack of fat, together with thin, wrinkled, loose skin and veins visible beneath the skin, makes affected individuals look older than their biological age. This appearance of premature aging is sometimes described as progeroid.
Affected individuals also have excessive hair growth (hypertrichosis) on their face and body. They have a low hairline on the forehead and their scalp hair is often coarse. People with Gorlin-Chaudhry-Moss syndrome also have shortened bones at the ends of the fingers and toes (short distal phalanges). Affected females have unusually small external genital folds (hypoplasia of the labia majora).
Some individuals with Gorlin-Chaudhry-Moss syndrome have mild developmental delay but intelligence is usually normal in this disorder, as is life expectancy.
Frequency
Gorlin-Chaudhry-Moss syndrome is an extremely rare condition. Approximately ten affected individuals have been described in the medical literature. All known individuals with this condition have been female.
Causes
Gorlin-Chaudhry-Moss syndrome can be caused by mutations in the SLC25A24 gene. This gene provides instructions for producing a protein that transports molecules across the inner membrane of the mitochondria, the energy-producing centers in cells. Among these molecules is ATP, which is the cell's main energy source. Transportation of ATP within the mitochondria is essential for normal energy production, the formation and breakdown (metabolism) of various molecules, and protein production within cells.
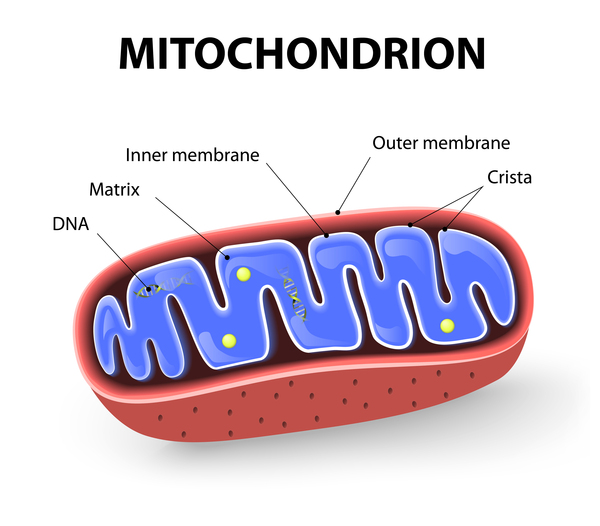
The mutations that cause Gorlin-Chaudhry-Moss syndrome are thought to alter the structure of the protein produced from the SLC25A24 gene, which likely decreases its ability to transport molecules across the mitochondrial inner membrane. As a result, there is an increase in mitochondrial size (mitochondria swelling), breakage of mitochondria into smaller pieces, and an overall decrease in energy production. This increase in abnormal mitochondria and decrease in cellular energy can lead to cell death. While altered cellular energy production and increased cell death are likely responsible for the features of Gorlin-Chaudhry-Moss syndrome, it is unclear how these changes lead to the specific signs and symptoms of the condition.
In some affected individuals, no mutation in the SLC25A24 gene has been found. Changes in other unknown genes may cause the disorder in these cases.
Inheritance
In individuals with an SLC25A24 gene mutation, Gorlin-Chaudhry-Moss syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In these cases, the condition results from new (de novo) mutations in the gene that occur during the formation of reproductive cells (eggs or sperm) or in early embryonic development.

Since all known individuals with Gorlin-Chaudhry-Moss syndrome have been female, there are likely other unknown, sex-related factors involved in the inheritance of this condition.
Other Names for This Condition
- Craniofacial dysostosis, hypertrichosis, hypoplasia of labia majora, dental and eye anomalies, patent ductus arteriosus, and normal intelligence
- Craniofacial dysostosis, patent ductus arteriosus, hypertrichosis, hypoplasia of labia majora, dental and eye anomalies
- GCM syndrome
- GCMS
- Gorlin Chaudhry Moss syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Aravena T, Passalacqua C, Pizarro O, Aracena M. Two sisters resembling Gorlin-Chaudhry-Moss syndrome. Am J Med Genet A. 2011 Oct;155A(10):2552-5. doi: 10.1002/ajmg.a.34204. Epub 2011 Sep 9. Citation on PubMed
- Ehmke N, Graul-Neumann L, Smorag L, Koenig R, Segebrecht L, Magoulas P, Scaglia F, Kilic E, Hennig AF, Adolphs N, Saha N, Fauler B, Kalscheuer VM, Hennig F, Altmuller J, Netzer C, Thiele H, Nurnberg P, Yigit G, Jager M, Hecht J, Kruger U, Mielke T, Krawitz PM, Horn D, Schuelke M, Mundlos S, Bacino CA, Bonnen PE, Wollnik B, Fischer-Zirnsak B, Kornak U. De Novo Mutations in SLC25A24 Cause a Craniosynostosis Syndrome with Hypertrichosis, Progeroid Appearance, and Mitochondrial Dysfunction. Am J Hum Genet. 2017 Nov 2;101(5):833-843. doi: 10.1016/j.ajhg.2017.09.016. Citation on PubMed or Free article on PubMed Central
- Rosti RO, Karaer K, Karaman B, Torun D, Guran S, Bahce M. Gorlin-Chaudhry-Moss syndrome revisited: expanding the phenotype. Am J Med Genet A. 2013 Jul;161A(7):1737-42. doi: 10.1002/ajmg.a.35954. Epub 2013 May 17. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.