

Description
Hypertrophic cardiomyopathy is a heart condition that is characterized by the thickening (hypertrophy) of the heart (cardiac) muscle. This condition is called nonsyndromic hypertrophic cardiomyopathy when it is not associated with signs and symptoms affecting other parts of the body and when it cannot be explained by other causes, such as chronic high blood pressure.
The symptoms of nonsyndromic hypertrophic cardiomyopathy often appear in adolescence or young adulthood, although they can begin at any time throughout life. In most affected individuals, hypertrophy occurs in the interventricular septum, which is the muscular wall that separates the lower left chamber of the heart (the left ventricle) from the lower right chamber (the right ventricle). This thickening of the interventricular septum can obstruct the flow of oxygen-rich blood from the heart, which may cause an abnormal heart sound during a heartbeat (heart murmur).
The features of nonsyndromic hypertrophic cardiomyopathy can vary, even among members of the same family. While some affected individuals have no associated symptoms, others may experience chest pain; shortness of breath, especially during physical activity; or a sensation of fluttering or pounding in the chest (palpitations). Affected individuals may also have episodes of dizziness or fainting (syncope).
Most people with nonsyndromic hypertrophic cardiomyopathy have a normal life expectancy. However, some affected individuals develop an abnormal heart rhythm (arrhythmia) that may be life-threatening. A small number of affected individuals develop heart failure. Although uncommon, people with nonsyndromic hypertrophic cardiomyopathy have an increased risk of sudden death, even if they have no other symptoms of the condition.
Frequency
Hypertrophic cardiomyopathy affects approximately 1 in 500 people worldwide. Nonsyndromic hypertrophic cardiomyopathy likely accounts for more than half of all cases.
Causes
Genetic changes that cause disease are called pathogenic variants. Pathogenic variants in one of several genes can cause nonsyndromic hypertrophic cardiomyopathy, but variants in the MYH7 and MYBPC3 genes are the most common genetic cause of this condition.
Many of the genes that are associated with nonsyndromic hypertrophic cardiomyopathy provide instructions for producing proteins that play an important role in the function of cardiac muscle. Many of these proteins are associated with structures called sarcomeres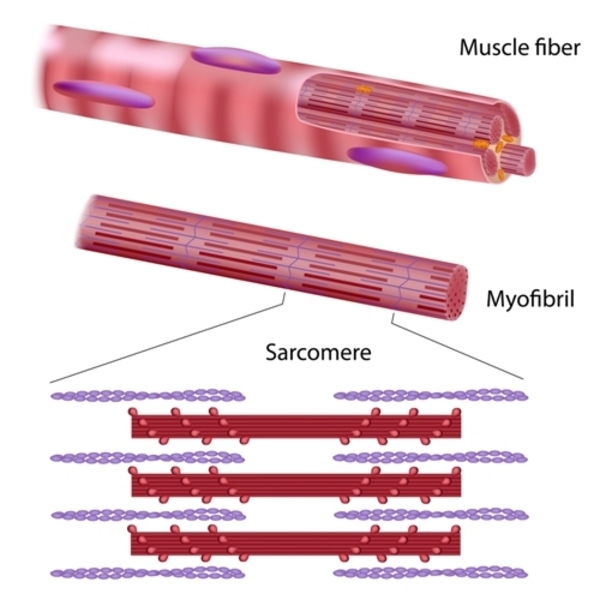 . Sarcomeres, which are the basic units of muscle contraction, are made up of overlapping thick and thin protein filaments
. Sarcomeres, which are the basic units of muscle contraction, are made up of overlapping thick and thin protein filaments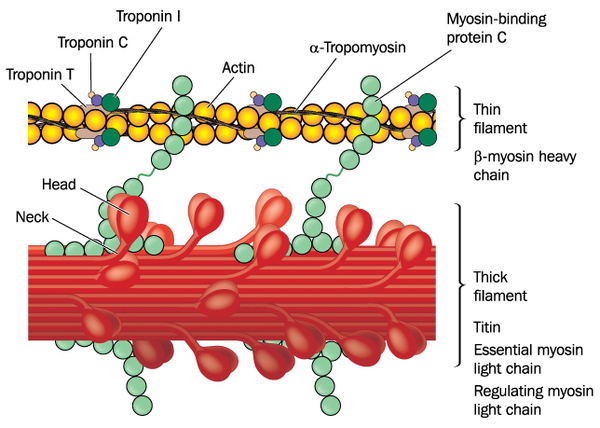 . These thick and thin filaments shift so that the thin filaments move past the thick filaments. This movement shortens the sarcomere and contracts the muscle. Regular contractions of cardiac muscle pump blood to the rest of the body.
. These thick and thin filaments shift so that the thin filaments move past the thick filaments. This movement shortens the sarcomere and contracts the muscle. Regular contractions of cardiac muscle pump blood to the rest of the body.
The protein produced from the MYH7 gene is the major component of the thick filament in sarcomeres. The protein produced from the MYBPC3 gene provides structural support to the thick filament and helps regulate muscle contractions.
Pathogenic variants in the genes that are associated with nonsyndromic hypertrophic cardiomyopathy cause cells to produce proteins that do not function properly. Many of these altered proteins impair the function of the sarcomeres and disrupt cardiac muscle contraction. Researchers do not understand exactly how changes in the genes that are associated with cardiac muscle and sarcomere function lead to the characteristic features of nonsyndromic hypertrophic cardiomyopathy.
Approximately 70 percent of people with nonsyndromic hypertrophic cardiomyopathy do not have a pathogenic variant in one of the genes that is associated with this condition. In these cases, the cause of the condition is unknown. People who have pathogenic variants in the genes that are associated with sarcomere function tend to show signs and symptoms of nonsyndromic hypertrophic cardiomyopathy earlier in life than people in whom no pathogenic variants are detected.
Hypertrophic cardiomyopathy may occur as part of a syndrome that affects other organs and tissues in the body. These forms of the condition are described as "syndromic" and are caused by variants in other genes.
Inheritance
Nonsyndromic hypertrophic cardiomyopathy has different inheritance patterns depending on the specific gene involved. When nonsyndromic hypertrophic cardiomyopathy occurs in multiple family members, it may be called familial hypertrophic cardiomyopathy.
Nonsyndromic hypertrophic cardiomyopathy is typically inherited in an autosomal dominant pattern , which means one copy of the altered gene in each cell is sufficient to cause the disorder. However, some people who have the altered gene never develop features of the condition. This is known as incomplete penetrance.
, which means one copy of the altered gene in each cell is sufficient to cause the disorder. However, some people who have the altered gene never develop features of the condition. This is known as incomplete penetrance.
Although many individuals with nonsyndromic hypertrophic cardiomyopathy have a parent with the condition, some people have the condition as a result of a new (de novo) variant in a gene that occurs during the formation of reproductive cells (eggs or sperm) in an individual's parent or during early embryonic development.
Some cases of nonsyndromic hypertrophic cardiomyopathy are inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a pathogenic variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a pathogenic variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
In rare cases, affected individuals have a pathogenic variant in two or more genes that are associated with the condition. This can lead to more severe signs and symptoms.
When hypertrophic cardiomyopathy is part of a syndrome, it follows the inheritance pattern of that syndrome.
Other Names for This Condition
- CMH
- Familial hypertrophic cardiomyopathy
- FHM
- HCM
- Hereditary ventricular hypertrophy
- Idiopathic hypertrophic subaortic stenosis
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Hypertrophic cardiomyopathy 1

- Genetic Testing Registry: Hypertrophic cardiomyopathy 10
- Genetic Testing Registry: Hypertrophic cardiomyopathy 11
- Genetic Testing Registry: Hypertrophic cardiomyopathy 12
- Genetic Testing Registry: Hypertrophic cardiomyopathy 13
- Genetic Testing Registry: Hypertrophic cardiomyopathy 17
- Genetic Testing Registry: Hypertrophic cardiomyopathy 18
- Genetic Testing Registry: Hypertrophic cardiomyopathy 2
- Genetic Testing Registry: Hypertrophic cardiomyopathy 3
- Genetic Testing Registry: Hypertrophic cardiomyopathy 4
- Genetic Testing Registry: Hypertrophic cardiomyopathy 7
- Genetic Testing Registry: Hypertrophic cardiomyopathy 8
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
- CARDIOMYOPATHY, FAMILIAL HYPERTROPHIC, 10; CMH10
- CARDIOMYOPATHY, FAMILIAL HYPERTROPHIC, 11; CMH11
- CARDIOMYOPATHY, FAMILIAL HYPERTROPHIC, 12; CMH12
- CARDIOMYOPATHY, FAMILIAL HYPERTROPHIC, 13; CMH13
- CARDIOMYOPATHY, FAMILIAL HYPERTROPHIC, 17; CMH17
- CARDIOMYOPATHY, FAMILIAL HYPERTROPHIC, 18; CMH18
- CARDIOMYOPATHY, FAMILIAL HYPERTROPHIC, 1; CMH1
- CARDIOMYOPATHY, FAMILIAL HYPERTROPHIC, 2; CMH2
- CARDIOMYOPATHY, FAMILIAL HYPERTROPHIC, 3; CMH3
- CARDIOMYOPATHY, FAMILIAL HYPERTROPHIC, 4; CMH4
- CARDIOMYOPATHY, FAMILIAL HYPERTROPHIC, 7; CMH7
- CARDIOMYOPATHY, FAMILIAL HYPERTROPHIC, 8; CMH8
Scientific Articles on PubMed
References
- Authors/Task Force members; Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, McKenna WJ, Mogensen J, Nihoyannopoulos P, Nistri S, Pieper PG, Pieske B, Rapezzi C, Rutten FH, Tillmanns C, Watkins H. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014 Oct 14;35(39):2733-79. doi: 10.1093/eurheartj/ehu284. Epub 2014 Aug 29. No abstract available. Citation on PubMed
- Bashyam MD, Savithri GR, Kumar MS, Narasimhan C, Nallari P. Molecular genetics of familial hypertrophic cardiomyopathy (FHC). J Hum Genet. 2003;48(2):55-64. doi: 10.1007/s100380300007. Citation on PubMed
- Basit H, Alahmadi MH, Sharma S. Hypertrophic Cardiomyopathy. 2024 Jun 7. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from http://www.ncbi.nlm.nih.gov/books/NBK430788/ Citation on PubMed
- Carrier L. Targeting the population for gene therapy with MYBPC3. J Mol Cell Cardiol. 2021 Jan;150:101-108. doi: 10.1016/j.yjmcc.2020.10.003. Epub 2020 Oct 11. Citation on PubMed
- Cirino AL, Channaoui N, Ho C. Nonsyndromic Hypertrophic Cardiomyopathy Overview. 2008 Aug 5 [updated 2025 Mar 6]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1768/ Citation on PubMed
- Frey N, Luedde M, Katus HA. Mechanisms of disease: hypertrophic cardiomyopathy. Nat Rev Cardiol. 2011 Oct 25;9(2):91-100. doi: 10.1038/nrcardio.2011.159. Citation on PubMed
- Hespe S, Waddell A, Asatryan B, Owens E, Thaxton C, Adduru ML, Anderson K, Brown EE, Hoffman-Andrews L, Jordan E, Josephs K, Mayers M, Peters S, Stafford F, Bagnall RD, Bronicki L, Callewaert B, Chahal CAA, James CA, Jarinova O, Landstrom AP, McNally EM, Murray B, Muino-Mosquera L, Parikh V, Reuter C, Walsh R, Wayburn B, Ware JS, Ingles J. Genes Associated With Hypertrophic Cardiomyopathy: A Reappraisal by the ClinGen Hereditary Cardiovascular Disease Gene Curation Expert Panel. J Am Coll Cardiol. 2025 Feb 25;85(7):727-740. doi: 10.1016/j.jacc.2024.12.010. Citation on PubMed
- Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, Cirino AL, Fox JC, Lakdawala NK, Ware JS, Caleshu CA, Helms AS, Colan SD, Girolami F, Cecchi F, Seidman CE, Sajeev G, Signorovitch J, Green EM, Olivotto I. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 2018 Oct 2;138(14):1387-1398. doi: 10.1161/CIRCULATIONAHA.117.033200. Epub 2018 Aug 23. Citation on PubMed
- Ho CY. New Paradigms in Hypertrophic Cardiomyopathy: Insights from Genetics. Prog Pediatr Cardiol. 2011 May;31(2):93-98. doi: 10.1016/j.ppedcard.2011.02.005. Citation on PubMed or Free article on PubMed Central
- Ingles J, Burns C, Bagnall RD, Lam L, Yeates L, Sarina T, Puranik R, Briffa T, Atherton JJ, Driscoll T, Semsarian C. Nonfamilial Hypertrophic Cardiomyopathy: Prevalence, Natural History, and Clinical Implications. Circ Cardiovasc Genet. 2017 Apr;10(2):e001620. doi: 10.1161/CIRCGENETICS.116.001620. Citation on PubMed
- Keren A, Syrris P, McKenna WJ. Hypertrophic cardiomyopathy: the genetic determinants of clinical disease expression. Nat Clin Pract Cardiovasc Med. 2008 Mar;5(3):158-68. doi: 10.1038/ncpcardio1110. Epub 2008 Jan 29. Citation on PubMed
- Kimura A. Molecular genetics and pathogenesis of cardiomyopathy. J Hum Genet. 2016 Jan;61(1):41-50. doi: 10.1038/jhg.2015.83. Epub 2015 Jul 16. Citation on PubMed
- Ko C, Arscott P, Concannon M, Saberi S, Day SM, Yashar BM, Helms AS. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med. 2018 Jan;20(1):69-75. doi: 10.1038/gim.2017.79. Epub 2017 Jun 22. Citation on PubMed
- Litt MJ, Ali A, Reza N. Familial Hypertrophic Cardiomyopathy: Diagnosis and Management. Vasc Health Risk Manag. 2023 Apr 6;19:211-221. doi: 10.2147/VHRM.S365001. eCollection 2023. Citation on PubMed
- Lopes LR, Ho CY, Elliott PM. Genetics of hypertrophic cardiomyopathy: established and emerging implications for clinical practice. Eur Heart J. 2024 Aug 9;45(30):2727-2734. doi: 10.1093/eurheartj/ehae421. Citation on PubMed
- Lopes LR, Macken WL, Preez SD, Kotwal H, Savvatis K, Sekhri N, Mohiddin SA, Kabiljo R, Pitceathly RDS. An analysis of mitochondrial variation in cardiomyopathy patients from the 100,000 genomes cohort: m.4300A>G as a cause of genetically elusive hypertrophic cardiomyopathy. Hum Genomics. 2024 Dec 5;18(1):136. doi: 10.1186/s40246-024-00702-9. Citation on PubMed
- Maron BA, Wang RS, Carnethon MR, Rowin EJ, Loscalzo J, Maron BJ, Maron MS. What Causes Hypertrophic Cardiomyopathy? Am J Cardiol. 2022 Sep 15;179:74-82. doi: 10.1016/j.amjcard.2022.06.017. Epub 2022 Jul 14. Citation on PubMed
- Marston S, Copeland O, Gehmlich K, Schlossarek S, Carrier L. How do MYBPC3 mutations cause hypertrophic cardiomyopathy? J Muscle Res Cell Motil. 2012 May;33(1):75-80. doi: 10.1007/s10974-011-9268-3. Epub 2011 Nov 5. Citation on PubMed
- Massera D, Sherrid MV, Maron MS, Rowin EJ, Maron BJ. How common is hypertrophic cardiomyopathy... really?: Disease prevalence revisited 27 years after CARDIA. Int J Cardiol. 2023 Jul 1;382:64-67. doi: 10.1016/j.ijcard.2023.04.005. Epub 2023 Apr 5. Citation on PubMed
- Rodriguez JE, McCudden CR, Willis MS. Familial hypertrophic cardiomyopathy: basic concepts and future molecular diagnostics. Clin Biochem. 2009 Jun;42(9):755-65. doi: 10.1016/j.clinbiochem.2009.01.020. Epub 2009 Feb 9. Citation on PubMed
- Toste A. Advances in hypertrophic cardiomyopathy: What the cardiologist needs to know. Rev Port Cardiol. 2022 Jun;41(6):499-509. doi: 10.1016/j.repc.2021.05.015. Epub 2022 May 27. English, Portuguese. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.