

Description
Familial hypercholesterolemia is an inherited condition characterized by very high levels of cholesterol in the blood. Cholesterol is a waxy, fat-like substance that is produced in the body and obtained from foods that come from animals (particularly egg yolks, meat, poultry, fish, and dairy products). The body needs this substance to build cell membranes, make certain hormones, and produce compounds that aid in fat digestion. In people with familial hypercholesterolemia, the body is unable to get rid of extra cholesterol, and it builds up in the blood. Too much cholesterol increases a person's risk of developing heart disease.
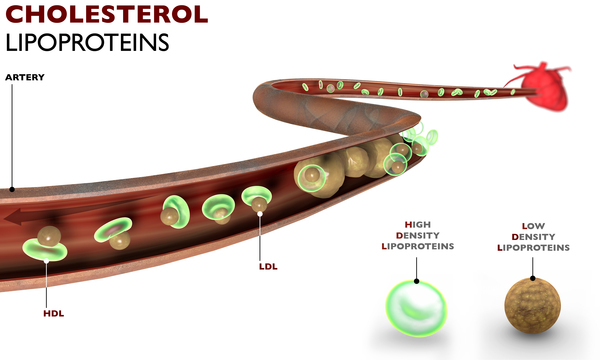
People with familial hypercholesterolemia have a high risk of developing a form of heart disease called coronary artery disease at a young age. This condition occurs when excess cholesterol in the bloodstream is deposited on the inner walls of blood vessels, particularly the arteries that supply blood to the heart (coronary arteries). The abnormal buildup of cholesterol forms clumps (plaques) that narrow and harden artery walls. As the plaques get bigger, they can clog the arteries and restrict the flow of blood to the heart. The buildup of plaques in coronary arteries causes a form of chest pain called angina and greatly increases a person's risk of having a heart attack.
Familial hypercholesterolemia can also cause health problems related to the buildup of excess cholesterol in tissues other than the heart and blood vessels. If cholesterol accumulates in the tissues that attach muscles to bones (tendons), it causes characteristic growths called tendon xanthomas. These growths most often affect the Achilles tendons, which attach the calf muscles to the heels, and tendons in the hands and fingers. Yellowish cholesterol deposits can develop under the skin of the eyelids and are known as xanthelasmata. Cholesterol can also accumulate at the edges of the clear, front surface of the eye (the cornea), leading to a gray-colored ring called an arcus cornealis.
Frequency
Familial hypercholesterolemia affects an estimated 1 in 200 to 1 in 250 people in most countries and is thought to be the most common inherited condition affecting the heart and blood vessels (cardiovascular disease). The condition occurs even more frequently in certain populations, including Afrikaners in South Africa, Lebanese, and Tunisians.
Causes
Mutations in the APOB, LDLR, LDLRAP1, or PCSK9 gene cause familial hypercholesterolemia. Changes in the LDLR gene are the most common cause of this condition. The LDLR gene provides instructions for making a protein called a low-density lipoprotein receptor. This type of receptor binds to particles called low-density lipoproteins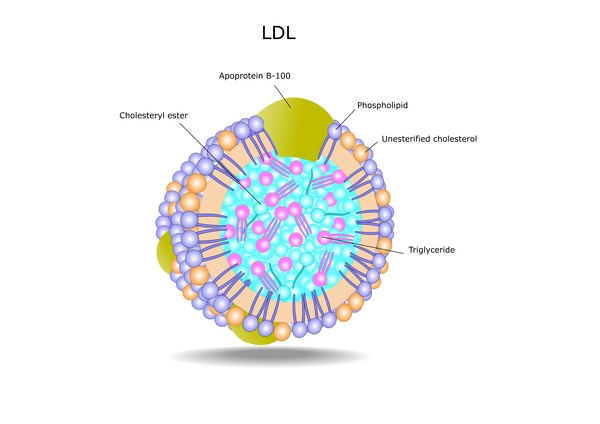 (LDLs), which are the primary carriers of cholesterol in the blood. By removing LDLs from the bloodstream, these receptors play a critical role in regulating cholesterol levels. Some LDLR gene mutations reduce the number of low-density lipoprotein receptors produced within cells. Other mutations disrupt the receptors' ability to remove low-density lipoproteins from the bloodstream. As a result, people with mutations in the LDLR gene have very high levels of blood cholesterol. As the excess cholesterol circulates through the bloodstream, it is deposited abnormally in tissues such as the skin, tendons, and coronary arteries.
(LDLs), which are the primary carriers of cholesterol in the blood. By removing LDLs from the bloodstream, these receptors play a critical role in regulating cholesterol levels. Some LDLR gene mutations reduce the number of low-density lipoprotein receptors produced within cells. Other mutations disrupt the receptors' ability to remove low-density lipoproteins from the bloodstream. As a result, people with mutations in the LDLR gene have very high levels of blood cholesterol. As the excess cholesterol circulates through the bloodstream, it is deposited abnormally in tissues such as the skin, tendons, and coronary arteries.
Less commonly, familial hypercholesterolemia is caused by mutations in the APOB, LDLRAP1, or PCSK9 gene. Proteins produced from these genes are essential for the normal function of low-density lipoprotein receptors. Mutations in any of these genes prevent cells from making functional receptors or alter the receptors' function. Hypercholesterolemia results when low-density lipoprotein receptors are unable to remove cholesterol from the blood effectively. Some people with familial hypercholesterolemia do not have a mutation in one of these genes. In these cases, the cause of the condition is unknown.
Both genetic and environmental risk factors play roles in familial hypercholesterolemia. Lifestyle choices including diet, exercise, and tobacco smoking strongly influence the amount of cholesterol in the blood and the risk of coronary artery disease. Additional factors that impact the outcome of the condition include a person's sex, age, and chronic diseases such as diabetes and obesity.
Familial hypercholesterolemia accounts for only a small percentage of all cases of high cholesterol. Researchers are working to identify and characterize additional genes that may influence cholesterol levels and the risk of heart disease in people with other forms of hypercholesterolemia.
Inheritance
Familial hypercholesterolemia resulting from mutations in the LDLR, APOB, or PCSK9 gene have an autosomal dominant pattern of inheritance. Autosomal dominant inheritance means one copy of an altered gene in each cell is sufficient to cause the disorder. An affected person typically inherits one altered copy of the gene from an affected parent and one normal copy of the gene from the other parent.

Rarely, a person with familial hypercholesterolemia has a mutation in both copies of the LDLR, APOB, or PCSK9 gene. This situation occurs when both parents have familial hypercholesterolemia, and each passes on one altered copy of the gene. The presence of two mutations results in a more severe form of familial hypercholesterolemia that usually appears in childhood.
When familial hypercholesterolemia is caused by mutations in the LDLRAP1 gene, the condition is inherited in an autosomal recessive pattern. Autosomal recessive inheritance means the condition results from two altered copies of the gene in each cell. The parents of an individual with autosomal recessive hypercholesterolemia each carry one copy of the altered gene, but their blood cholesterol levels are usually in the normal range.

Other Names for This Condition
- Familial hypercholesterolaemia
- FH
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Austin MA, Hutter CM, Zimmern RL, Humphries SE. Familial hypercholesterolemia and coronary heart disease: a HuGE association review. Am J Epidemiol. 2004 Sep 1;160(5):421-9. doi: 10.1093/aje/kwh237. Citation on PubMed
- Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol. 2004 Sep 1;160(5):407-20. doi: 10.1093/aje/kwh236. Citation on PubMed
- Civeira F; International Panel on Management of Familial Hypercholesterolemia. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis. 2004 Mar;173(1):55-68. doi: 10.1016/j.atherosclerosis.2003.11.010. Citation on PubMed
- Defesche JC, Gidding SS, Harada-Shiba M, Hegele RA, Santos RD, Wierzbicki AS. Familial hypercholesterolaemia. Nat Rev Dis Primers. 2017 Dec 7;3:17093. doi: 10.1038/nrdp.2017.93. Citation on PubMed
- Ison HE, Clarke SL, Knowles JW. Familial Hypercholesterolemia. 2014 Jan 2 [updated 2025 Jan 30]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK174884/ Citation on PubMed
- Marais AD, Firth JC, Blom DJ. Homozygous familial hypercholesterolemia and its management. Semin Vasc Med. 2004 Feb;4(1):43-50. doi: 10.1055/s-2004-822985. Citation on PubMed
- Paththinige CS, Sirisena ND, Dissanayake V. Genetic determinants of inherited susceptibility to hypercholesterolemia - a comprehensive literature review. Lipids Health Dis. 2017 Jun 2;16(1):103. doi: 10.1186/s12944-017-0488-4. Citation on PubMed or Free article on PubMed Central
- Soutar AK, Naoumova RP. Autosomal recessive hypercholesterolemia. Semin Vasc Med. 2004 Aug;4(3):241-8. doi: 10.1055/s-2004-861491. Citation on PubMed
- van Aalst-Cohen ES, Jansen AC, de Jongh S, de Sauvage Nolting PR, Kastelein JJ. Clinical, diagnostic, and therapeutic aspects of familial hypercholesterolemia. Semin Vasc Med. 2004 Feb;4(1):31-41. doi: 10.1055/s-2004-822984. Citation on PubMed
- Yuan G, Wang J, Hegele RA. Heterozygous familial hypercholesterolemia: an underrecognized cause of early cardiovascular disease. CMAJ. 2006 Apr 11;174(8):1124-9. doi: 10.1503/cmaj.051313. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.