

Description
Factor XI deficiency is a disorder that can cause abnormal bleeding due to a shortage (deficiency) of the factor XI protein, which is involved in blood clotting. This condition is classified as either partial or severe based on the degree of deficiency of the factor XI protein. However, regardless of the severity of the protein deficiency, most affected individuals have relatively mild bleeding problems, and some people with this disorder have few if any symptoms. The most common feature of factor XI deficiency is prolonged bleeding after trauma or surgery, especially involving the inside of the mouth and nose (oral and nasal cavities) or the urinary tract. If the bleeding is left untreated after surgery, solid swellings consisting of congealed blood (hematomas) can develop in the surgical area.

Other signs and symptoms of this disorder can include frequent nosebleeds, easy bruising, bleeding under the skin, and bleeding of the gums. Women with this disorder can have heavy or prolonged menstrual bleeding (menorrhagia) or prolonged bleeding after childbirth. In contrast to some other bleeding disorders, spontaneous bleeding into the urine (hematuria), gastrointestinal tract, or skull cavity are not common in factor XI deficiency, although they can occur in severely affected individuals. Bleeding into the muscles or joints, which can cause long-term disability in other bleeding disorders, generally does not occur in this condition.
Frequency
Factor XI deficiency is estimated to affect approximately 1 in 1 million people worldwide. The severe deficiency disorder is much more common in people with central and eastern European (Ashkenazi) Jewish ancestry, occurring in about 1 in 450 individuals in that population. Researchers suggest that the actual prevalence of factor XI deficiency may be higher than reported, because mild cases of the disorder often do not come to medical attention.
Causes
Most cases of factor XI deficiency are caused by mutations in the F11 gene, which provides instructions for making the factor XI protein. This protein plays a role in the coagulation cascade, which is a series of chemical reactions that forms blood clots in response to injury. After an injury, clots seal off blood vessels to stop bleeding and trigger blood vessel repair.
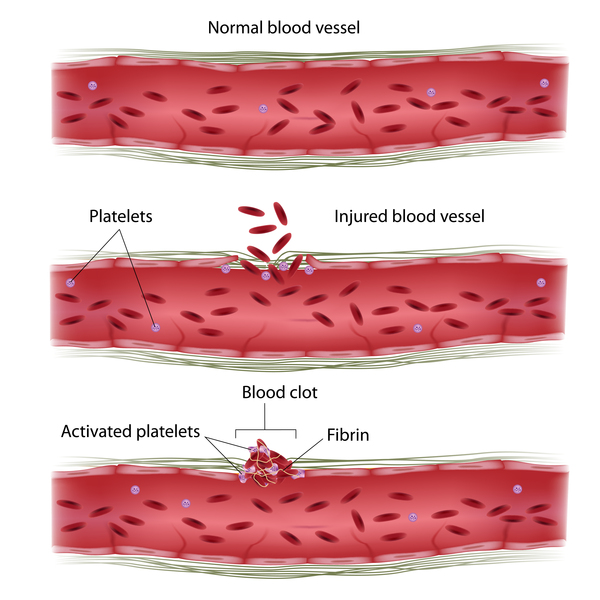
Mutations in the F11 gene result in a shortage (deficiency) of functional factor XI. This deficiency impairs the coagulation cascade, slowing the process of blood clotting and leading to the bleeding problems associated with this disorder. The amount of functional factor XI remaining varies depending on the particular mutation and whether one or both copies of the F11 gene in each cell have mutations. However, the severity of the bleeding problems in affected individuals does not necessarily correspond to the amount of factor XI in the bloodstream, and can vary even within the same family. Other genetic and environmental factors likely play a role in determining the severity of this condition.
Some cases of factor XI deficiency are not caused by F11 gene mutations. In these cases, the condition is called acquired factor XI deficiency. It can be caused by other disorders such as conditions in which the immune system malfunctions and attacks the factor XI protein. Because factor XI is made primarily by cells in the liver, acquired factor XI deficiency can also occur as the result of severe liver disease or receiving a transplanted liver from an affected individual. In addition, approximately 25 percent of people with another disorder called Noonan syndrome have factor XI deficiency.

Inheritance
Severe factor XI deficiency is passed down in an autosomal recessive pattern, which means both copies of the F11 gene in each cell have mutations. The parents of these individuals each carry one copy of the mutated gene and have partial factor XI deficiency; they rarely show severe signs and symptoms of the condition.

In some families, this condition is inherited in an autosomal dominant pattern, which means one copy of the altered F11 gene in each cell is sufficient to cause the disorder. In these cases, an affected person has one parent with the condition.

The acquired form of factor XI deficiency is not inherited and does not run in families.
Other Names for This Condition
- F11 deficiency
- Factor 11 deficiency
- Haemophilia C
- Hemophilia C
- Plasma thromboplastin antecedent deficiency
- PTA deficiency
- Rosenthal factor deficiency
- Rosenthal syndrome
- Rosenthal's disease
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Duga S, Salomon O. Congenital factor XI deficiency: an update. Semin Thromb Hemost. 2013 Sep;39(6):621-31. doi: 10.1055/s-0033-1353420. Epub 2013 Aug 8. Citation on PubMed
- Duga S, Salomon O. Factor XI Deficiency. Semin Thromb Hemost. 2009 Jun;35(4):416-25. doi: 10.1055/s-0029-1225764. Epub 2009 Jul 13. Citation on PubMed
- Emsley J, McEwan PA, Gailani D. Structure and function of factor XI. Blood. 2010 Apr 1;115(13):2569-77. doi: 10.1182/blood-2009-09-199182. Epub 2010 Jan 28. Citation on PubMed or Free article on PubMed Central
- Gomez K, Bolton-Maggs P. Factor XI deficiency. Haemophilia. 2008 Nov;14(6):1183-9. doi: 10.1111/j.1365-2516.2008.01667.x. Epub 2008 Feb 27. Citation on PubMed
- He R, Chen D, He S. Factor XI: hemostasis, thrombosis, and antithrombosis. Thromb Res. 2012 May;129(5):541-50. doi: 10.1016/j.thromres.2011.11.051. Epub 2011 Dec 22. Citation on PubMed
- Puy C, Rigg RA, McCarty OJ. The hemostatic role of factor XI. Thromb Res. 2016 May;141 Suppl 2(Suppl 2):S8-S11. doi: 10.1016/S0049-3848(16)30354-1. Citation on PubMed
- Wheeler AP, Gailani D. Why factor XI deficiency is a clinical concern. Expert Rev Hematol. 2016 Jul;9(7):629-37. doi: 10.1080/17474086.2016.1191944. Epub 2016 Jun 24. Citation on PubMed or Free article on PubMed Central
- Yankol Y, Mecit N, Kanmaz T, Acarli K, Kalayoglu M. Acquired factor XI deficiency: a rare complication after liver transplantation. Transplant Proc. 2015 Jan-Feb;47(1):179-81. doi: 10.1016/j.transproceed.2014.10.042. Epub 2015 Jan 14. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.