

Description
Noonan syndrome is a condition that affects many areas of the body. It is characterized by mildly unusual facial features, short stature, heart defects, bleeding problems, skeletal malformations, and many other signs and symptoms.
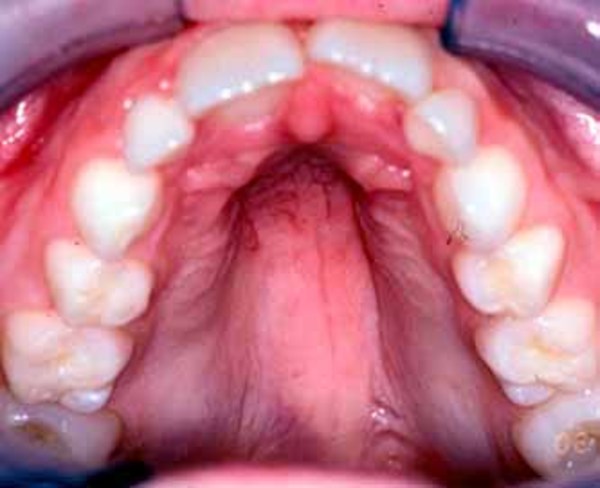
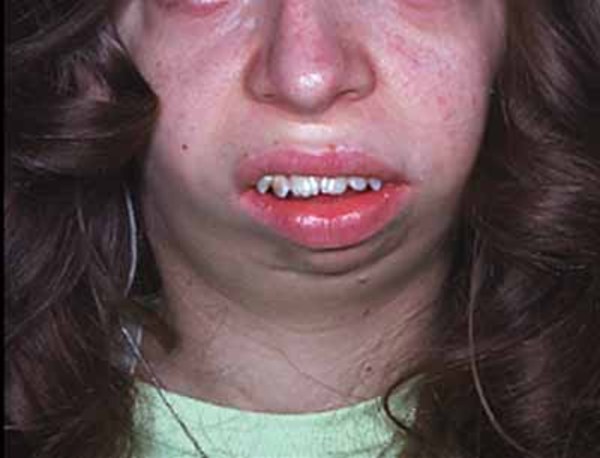
People with Noonan syndrome have distinctive facial features such as a deep groove in the area between the nose and mouth (philtrum), widely spaced eyes that are usually pale blue or blue-green in color, and low-set ears that are rotated backward. Affected individuals may have a high arch in the roof of the mouth (high-arched palate), poor teeth alignment, and a small lower jaw (micrognathia). Many children with Noonan syndrome have a short neck, and both children and adults may have excess neck skin (also called webbing) and a low hairline at the back of the neck.
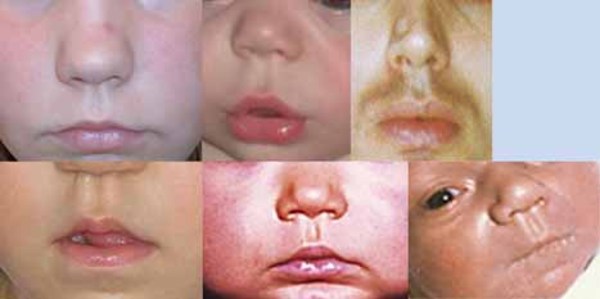

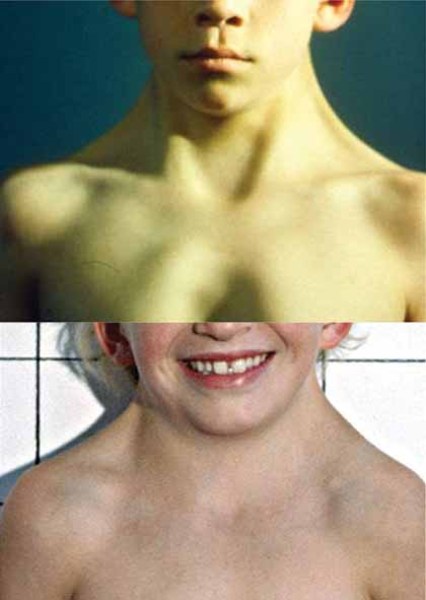
Between 50 and 70 percent of individuals with Noonan syndrome have short stature. At birth, they are usually a normal length and weight, but growth slows over time. Abnormal levels of growth hormone, a protein that is necessary for the normal growth of the body's bones and tissues, may contribute to the slow growth.
Individuals with Noonan syndrome often have either a sunken chest (pectus excavatum) or a protruding chest (pectus carinatum). Some affected people may also have an abnormal side-to-side curvature of the spine (scoliosis).

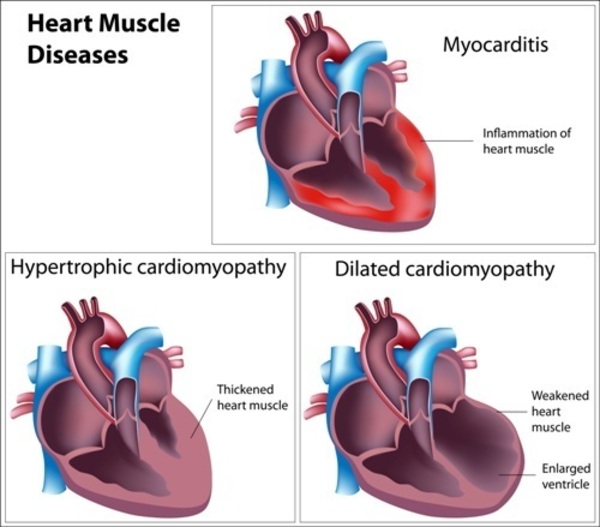
Most people with Noonan syndrome have some form of critical congenital heart disease. The most common heart defect in these individuals is a narrowing of the valve that controls blood flow from the heart to the lungs (pulmonary valve stenosis). Some have hypertrophic cardiomyopathy, which enlarges and weakens the heart muscle.
A variety of bleeding disorders have been associated with Noonan syndrome. Some affected individuals have excessive bruising, nosebleeds, or prolonged bleeding following injury or surgery. Rarely, women with Noonan syndrome who have a bleeding disorder have excessive bleeding during menstruation (menorrhagia) or childbirth.
Adolescent males with Noonan syndrome typically experience delayed puberty. They go through puberty starting at age 13 or 14 and have a reduced pubertal growth spurt that results in shortened stature. Most males with Noonan syndrome have undescended testes (cryptorchidism), which may contribute to infertility (inability to father a child) later in life. Females with Noonan syndrome can experience delayed puberty but most have normal puberty and fertility.
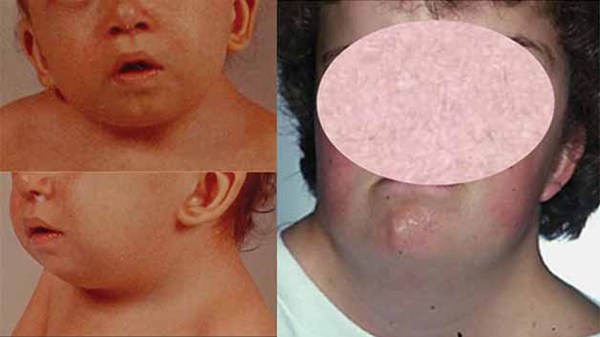
Noonan syndrome can cause a variety of other signs and symptoms. Most children diagnosed with Noonan syndrome have normal intelligence, but a few have special educational needs, and some have intellectual disability. Some affected individuals have vision or hearing problems. Affected infants may have feeding problems, which typically get better by age 1 or 2 years. Infants with Noonan syndrome may be born with puffy hands and feet caused by a buildup of fluid (lymphedema), which can go away on its own. Older individuals can also develop lymphedema, usually in the ankles and lower legs.
Some people with Noonan syndrome develop cancer, particularly those involving the blood-forming cells (leukemia). It has been estimated that children with Noonan syndrome have an eightfold increased risk of developing leukemia or other cancers over age-matched peers.
Noonan syndrome is one of a group of related conditions, collectively known as RASopathies. These conditions all have similar signs and symptoms and are caused by changes in the same cell signaling pathway. In addition to Noonan syndrome, the RASopathies include cardiofaciocutaneous syndrome, Costello syndrome, neurofibromatosis type 1, Legius syndrome, and Noonan syndrome with multiple lentigines.
Frequency
Noonan syndrome occurs in approximately 1 in 1,000 to 2,500 people.
Causes
Mutations in multiple genes can cause Noonan syndrome. Mutations in the PTPN11 gene cause about half of all cases. SOS1 gene mutations cause an additional 10 to 15 percent, and RAF1 and RIT1 genes each account for about 5 percent of cases. Mutations in other genes each account for a small number of cases. The cause of Noonan syndrome in 15 to 20 percent of people with this disorder is unknown.
The PTPN11, SOS1, RAF1, and RIT1 genes all provide instructions for making proteins that are important in the RAS/MAPK cell signaling pathway, which is needed for cell division and growth (proliferation), the process by which cells mature to carry out specific functions (differentiation), and cell movement (migration). Many of the mutations in the genes associated with Noonan syndrome cause the resulting protein to be turned on (active) longer than normal, rather than promptly switching on and off in response to cell signals. This prolonged activation alters normal RAS/MAPK signaling, which disrupts the regulation of cell growth and division, leading to the characteristic features of Noonan syndrome.
Rarely, Noonan syndrome is associated with genes that are not involved in the RAS/MAPK cell signaling pathway. Researchers are working to determine how mutations in these genes can lead to the signs and symptoms of Noonan syndrome.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Other Names for This Condition
- Familial Turner syndrome
- Female pseudo-Turner syndrome
- Male Turner syndrome
- Noonan's syndrome
- Noonan-Ehmke syndrome
- NS
- Pseudo-Ullrich-Turner syndrome
- Turner phenotype with normal karyotype
- Turner syndrome in female with X chromosome
- Turner-like syndrome
- Ullrich-Noonan syndrome
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Noonan syndrome

- Genetic Testing Registry: Noonan syndrome 1
- Genetic Testing Registry: Noonan syndrome 10
- Genetic Testing Registry: Noonan syndrome 2
- Genetic Testing Registry: Noonan syndrome 3
- Genetic Testing Registry: Noonan syndrome 4
- Genetic Testing Registry: Noonan syndrome 5
- Genetic Testing Registry: Noonan syndrome 6
- Genetic Testing Registry: Noonan syndrome 7
- Genetic Testing Registry: Noonan syndrome 8
- Genetic Testing Registry: Noonan syndrome 9
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Scientific Articles on PubMed
References
- Chen PC, Yin J, Yu HW, Yuan T, Fernandez M, Yung CK, Trinh QM, Peltekova VD, Reid JG, Tworog-Dube E, Morgan MB, Muzny DM, Stein L, McPherson JD, Roberts AE, Gibbs RA, Neel BG, Kucherlapati R. Next-generation sequencing identifies rare variants associated with Noonan syndrome. Proc Natl Acad Sci U S A. 2014 Aug 5;111(31):11473-8. doi: 10.1073/pnas.1324128111. Epub 2014 Jul 21. Citation on PubMed or Free article on PubMed Central
- Cordeddu V, Yin JC, Gunnarsson C, Virtanen C, Drunat S, Lepri F, De Luca A, Rossi C, Ciolfi A, Pugh TJ, Bruselles A, Priest JR, Pennacchio LA, Lu Z, Danesh A, Quevedo R, Hamid A, Martinelli S, Pantaleoni F, Gnazzo M, Daniele P, Lissewski C, Bocchinfuso G, Stella L, Odent S, Philip N, Faivre L, Vlckova M, Seemanova E, Digilio C, Zenker M, Zampino G, Verloes A, Dallapiccola B, Roberts AE, Cave H, Gelb BD, Neel BG, Tartaglia M. Activating Mutations Affecting the Dbl Homology Domain of SOS2 Cause Noonan Syndrome. Hum Mutat. 2015 Nov;36(11):1080-7. doi: 10.1002/humu.22834. Epub 2015 Aug 3. Citation on PubMed or Free article on PubMed Central
- Flex E, Jaiswal M, Pantaleoni F, Martinelli S, Strullu M, Fansa EK, Caye A, De Luca A, Lepri F, Dvorsky R, Pannone L, Paolacci S, Zhang SC, Fodale V, Bocchinfuso G, Rossi C, Burkitt-Wright EM, Farrotti A, Stellacci E, Cecchetti S, Ferese R, Bottero L, Castro S, Fenneteau O, Brethon B, Sanchez M, Roberts AE, Yntema HG, Van Der Burgt I, Cianci P, Bondeson ML, Cristina Digilio M, Zampino G, Kerr B, Aoki Y, Loh ML, Palleschi A, Di Schiavi E, Care A, Selicorni A, Dallapiccola B, Cirstea IC, Stella L, Zenker M, Gelb BD, Cave H, Ahmadian MR, Tartaglia M. Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Hum Mol Genet. 2014 Aug 15;23(16):4315-27. doi: 10.1093/hmg/ddu148. Epub 2014 Apr 4. Citation on PubMed or Free article on PubMed Central
- Kouz K, Lissewski C, Spranger S, Mitter D, Riess A, Lopez-Gonzalez V, Luttgen S, Aydin H, von Deimling F, Evers C, Hahn A, Hempel M, Issa U, Kahlert AK, Lieb A, Villavicencio-Lorini P, Ballesta-Martinez MJ, Nampoothiri S, Ovens-Raeder A, Puchmajerova A, Satanovskij R, Seidel H, Unkelbach S, Zabel B, Kutsche K, Zenker M. Genotype and phenotype in patients with Noonan syndrome and a RIT1 mutation. Genet Med. 2016 Dec;18(12):1226-1234. doi: 10.1038/gim.2016.32. Epub 2016 Apr 21. Citation on PubMed
- Kratz CP, Franke L, Peters H, Kohlschmidt N, Kazmierczak B, Finckh U, Bier A, Eichhorn B, Blank C, Kraus C, Kohlhase J, Pauli S, Wildhardt G, Kutsche K, Auber B, Christmann A, Bachmann N, Mitter D, Cremer FW, Mayer K, Daumer-Haas C, Nevinny-Stickel-Hinzpeter C, Oeffner F, Schluter G, Gencik M, Uberlacker B, Lissewski C, Schanze I, Greene MH, Spix C, Zenker M. Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br J Cancer. 2015 Apr 14;112(8):1392-7. doi: 10.1038/bjc.2015.75. Epub 2015 Mar 5. Citation on PubMed or Free article on PubMed Central
- Milosavljevic D, Overwater E, Tamminga S, de Boer K, Elting MW, van Hoorn ME, Rinne T, Houweling AC. Two cases of RIT1 associated Noonan syndrome: Further delineation of the clinical phenotype and review of the literature. Am J Med Genet A. 2016 Jul;170(7):1874-80. doi: 10.1002/ajmg.a.37657. Epub 2016 Apr 25. Citation on PubMed
- Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013 Jan 26;381(9863):333-42. doi: 10.1016/S0140-6736(12)61023-X. Epub 2013 Jan 10. Citation on PubMed or Free article on PubMed Central
- Rohrer T. Noonan syndrome: introduction and basic clinical features. Horm Res. 2009 Dec;72 Suppl 2:3-7. doi: 10.1159/000243772. Epub 2009 Dec 22. Citation on PubMed
- Romano AA, Allanson JE, Dahlgren J, Gelb BD, Hall B, Pierpont ME, Roberts AE, Robinson W, Takemoto CM, Noonan JA. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics. 2010 Oct;126(4):746-59. doi: 10.1542/peds.2009-3207. Epub 2010 Sep 27. Citation on PubMed
- Schubbert S, Zenker M, Rowe SL, Boll S, Klein C, Bollag G, van der Burgt I, Musante L, Kalscheuer V, Wehner LE, Nguyen H, West B, Zhang KY, Sistermans E, Rauch A, Niemeyer CM, Shannon K, Kratz CP. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006 Mar;38(3):331-6. doi: 10.1038/ng1748. Epub 2006 Feb 12. Citation on PubMed
- Sznajer Y, Keren B, Baumann C, Pereira S, Alberti C, Elion J, Cave H, Verloes A. The spectrum of cardiac anomalies in Noonan syndrome as a result of mutations in the PTPN11 gene. Pediatrics. 2007 Jun;119(6):e1325-31. doi: 10.1542/peds.2006-0211. Epub 2007 May 21. Citation on PubMed
- Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A, Pandit B, Oishi K, Martinelli S, Schackwitz W, Ustaszewska A, Martin J, Bristow J, Carta C, Lepri F, Neri C, Vasta I, Gibson K, Curry CJ, Siguero JP, Digilio MC, Zampino G, Dallapiccola B, Bar-Sagi D, Gelb BD. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007 Jan;39(1):75-9. doi: 10.1038/ng1939. Epub 2006 Dec 13. Citation on PubMed
- van der Burgt I. Noonan syndrome. Orphanet J Rare Dis. 2007 Jan 14;2:4. doi: 10.1186/1750-1172-2-4. Citation on PubMed or Free article on PubMed Central
- Vissers LE, Bonetti M, Paardekooper Overman J, Nillesen WM, Frints SG, de Ligt J, Zampino G, Justino A, Machado JC, Schepens M, Brunner HG, Veltman JA, Scheffer H, Gros P, Costa JL, Tartaglia M, van der Burgt I, Yntema HG, den Hertog J. Heterozygous germline mutations in A2ML1 are associated with a disorder clinically related to Noonan syndrome. Eur J Hum Genet. 2015 Mar;23(3):317-24. doi: 10.1038/ejhg.2014.115. Epub 2014 Jun 18. Citation on PubMed or Free article on PubMed Central
- Yamamoto GL, Aguena M, Gos M, Hung C, Pilch J, Fahiminiya S, Abramowicz A, Cristian I, Buscarilli M, Naslavsky MS, Malaquias AC, Zatz M, Bodamer O, Majewski J, Jorge AA, Pereira AC, Kim CA, Passos-Bueno MR, Bertola DR. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J Med Genet. 2015 Jun;52(6):413-21. doi: 10.1136/jmedgenet-2015-103018. Epub 2015 Mar 20. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.