

Description
Donnai-Barrow syndrome is an inherited disorder that affects many parts of the body. This disorder is characterized by unusual facial features, including prominent, wide-set eyes with outer corners that point downward; a short bulbous nose with a flat nasal bridge; ears that are rotated backward; and a widow's peak hairline.
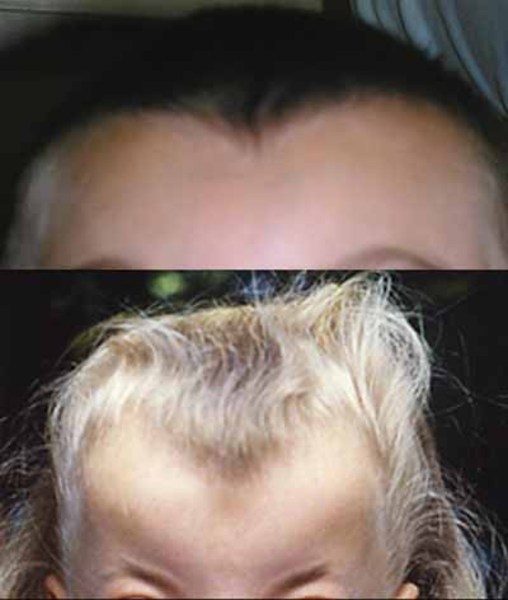
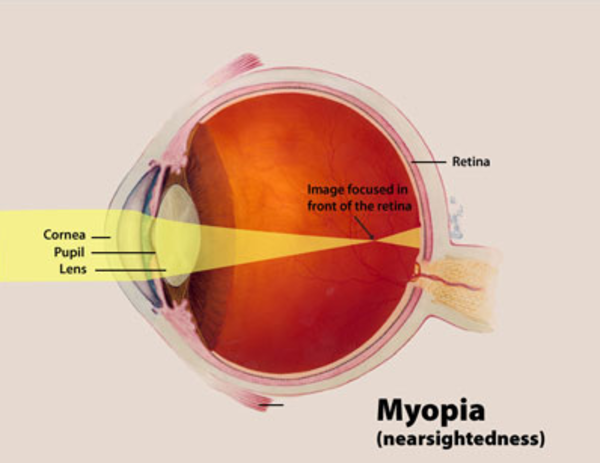
Individuals with Donnai-Barrow syndrome have severe hearing loss caused by abnormalities of the inner ear (sensorineural hearing loss). In addition, they often experience vision problems, including extreme nearsightedness (high myopia), detachment or deterioration of the light-sensitive tissue in the back of the eye (the retina), and progressive vision loss. Some have a gap or split in the colored part of the eye (iris coloboma).


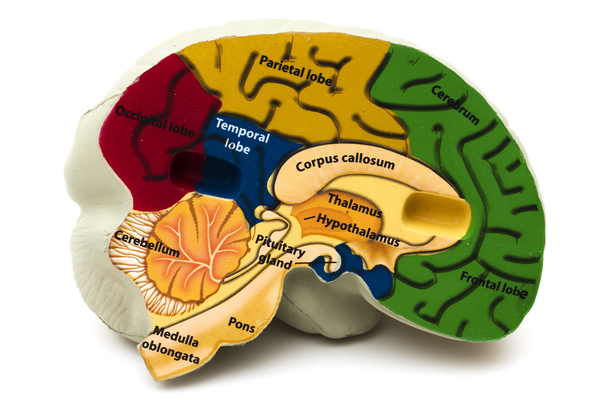
In almost all people with Donnai-Barrow syndrome, the tissue connecting the left and right halves of the brain (corpus callosum) is underdeveloped or absent. Affected individuals may also have other structural abnormalities of the brain. They generally have mild to moderate intellectual disability and developmental delay.
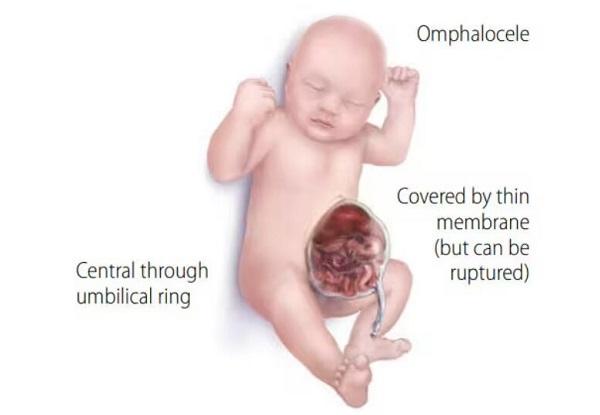
People with Donnai-Barrow syndrome may also have a hole in the muscle that separates the abdomen from the chest cavity (the diaphragm), which is called a congenital diaphragmatic hernia. This potentially serious birth defect allows the stomach and intestines to move into the chest and possibly crowd the developing heart and lungs. An opening in the wall of the abdomen (an omphalocele) that allows the abdominal organs to protrude through the navel may also occur in affected individuals. Occasionally people with Donnai-Barrow syndrome have abnormalities of the intestine, heart, or other organs.
Frequency
Although its prevalence is unknown, Donnai-Barrow syndrome appears to be a rare disorder. A few dozen affected individuals have been reported in many regions of the world.
Causes
Mutations in the LRP2 gene cause Donnai-Barrow syndrome. The LRP2 gene provides instructions for making a protein called megalin, which functions as a receptor. Receptor proteins have specific sites into which certain other proteins, called ligands, fit like keys into locks. Together, ligands and their receptors trigger signals that affect cell development and function. Megalin has many ligands involved in various body processes, including the absorption of vitamins A and D, immune functioning, stress response, and the transport of fats in the bloodstream.
Megalin is embedded in the membrane of cells that line the surfaces and cavities of the body (epithelial cells). The receptor helps move its ligands from the cell surface into the cell (endocytosis). It is active in the development and function of many parts of the body, including the brain and spinal cord (central nervous system), eyes, ears, lungs, intestine, reproductive system, and the small tubes in the kidneys where urine is formed (renal tubules).
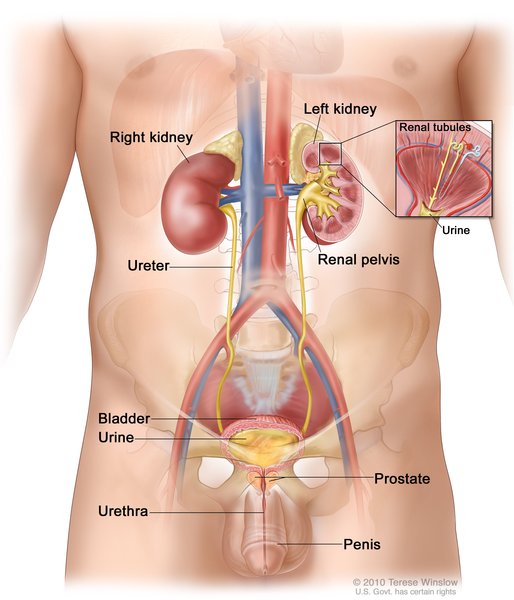
LRP2 gene mutations that cause Donnai-Barrow syndrome are believed to result in the absence of functional megalin protein. The lack of functional megalin in the renal tubules causes megalin's various ligands to be excreted in the urine rather than being absorbed back into the bloodstream. The features of Donnai-Barrow syndrome are probably caused by the inability of megalin to help absorb these ligands, disruption of biochemical signaling pathways, or other effects of the nonfunctional megalin protein. However, it is unclear how these abnormalities result in the specific signs and symptoms of the disorder.
A condition previously classified as a separate disorder called facio-oculo-acoustico-renal (FOAR) syndrome has also been found to be caused by LRP2 mutations. FOAR syndrome is now considered to be the same disorder as Donnai-Barrow syndrome.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. In almost all cases, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene but typically do not show signs and symptoms of the condition.

One individual with Donnai-Barrow syndrome was found to have inherited both copies of the mutated gene from his father as a result of a genetic change called uniparental disomy (UPD). UPD occurs when a person receives two copies of a chromosome, or part of a chromosome, from one parent and no copies from the other parent. UPD can occur as a random event during the formation of egg or sperm cells or may happen in early fetal development.

Other Names for This Condition
- DBS
- DBS/FOAR syndrome
- Diaphragmatic hernia-exomphalos-corpus callosum agenesis
- Diaphragmatic hernia-exomphalos-hypertelorism syndrome
- Faciooculoacousticorenal syndrome
- FOAR syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Chassaing N, Lacombe D, Carles D, Calvas P, Saura R, Bieth E. Donnai-Barrow syndrome: four additional patients. Am J Med Genet A. 2003 Sep 1;121A(3):258-62. doi: 10.1002/ajmg.a.20266. Citation on PubMed
- Chen CP. Syndromes and disorders associated with omphalocele (III): single gene disorders, neural tube defects, diaphragmatic defects and others. Taiwan J Obstet Gynecol. 2007 Jun;46(2):111-20. doi: 10.1016/S1028-4559(07)60004-7. Citation on PubMed
- Kantarci S, Al-Gazali L, Hill RS, Donnai D, Black GC, Bieth E, Chassaing N, Lacombe D, Devriendt K, Teebi A, Loscertales M, Robson C, Liu T, MacLaughlin DT, Noonan KM, Russell MK, Walsh CA, Donahoe PK, Pober BR. Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes. Nat Genet. 2007 Aug;39(8):957-9. doi: 10.1038/ng2063. Epub 2007 Jul 15. Citation on PubMed or Free article on PubMed Central
- Kantarci S, Ragge NK, Thomas NS, Robinson DO, Noonan KM, Russell MK, Donnai D, Raymond FL, Walsh CA, Donahoe PK, Pober BR. Donnai-Barrow syndrome (DBS/FOAR) in a child with a homozygous LRP2 mutation due to complete chromosome 2 paternal isodisomy. Am J Med Genet A. 2008 Jul 15;146A(14):1842-7. doi: 10.1002/ajmg.a.32381. Citation on PubMed or Free article on PubMed Central
- Longoni M, Kantarci S, Donnai D, Pober BR. Donnai-Barrow Syndrome. 2008 Aug 28 [updated 2018 Nov 21]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1878/ Citation on PubMed
- Patel N, Hejkal T, Katz A, Margalit E. Ocular manifestations of Donnai-Barrow syndrome. J Child Neurol. 2007 Apr;22(4):462-4. doi: 10.1177/0883073807301933. Citation on PubMed
- Pober BR, Longoni M, Noonan KM. A review of Donnai-Barrow and facio-oculo-acoustico-renal (DB/FOAR) syndrome: clinical features and differential diagnosis. Birth Defects Res A Clin Mol Teratol. 2009 Jan;85(1):76-81. doi: 10.1002/bdra.20534. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.