

Description
Cholangiocarcinoma is a group of cancers that begin in the bile ducts. Bile ducts are branched tubes that connect the liver and gallbladder to the small intestine. They carry bile, which is a fluid that helps the body digest fats that are in food. Bile is made in the liver and stored in the gallbladder before being released in the small intestine after a person eats.


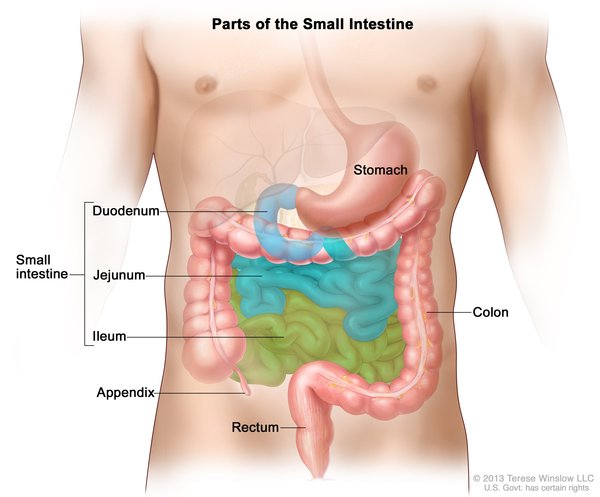
Cholangiocarcinoma is classified by its location in relation to the liver. Intrahepatic cholangiocarcinoma begins in the small bile ducts within the liver. This is the least common form of the disease, accounting for less than 10 percent of all cases. Perihilar cholangiocarcinoma (also known as a Klatskin tumor) begins in an area called the hilum, where the right and left major bile ducts join and leave the liver. It is the most common form of the disease, accounting for more than half of all cases. The remaining cases are classified as distal cholangiocarcinomas, which begin in bile ducts outside the liver. The perihilar and distal forms of the disease, which both occur outside the liver, are sometimes grouped together and called extrahepatic cholangiocarcinoma.
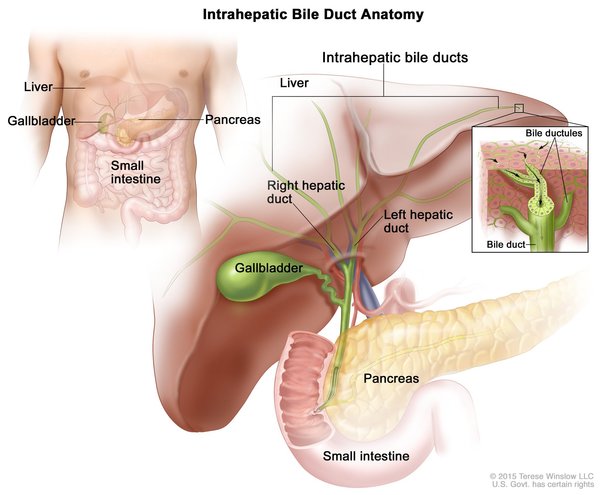
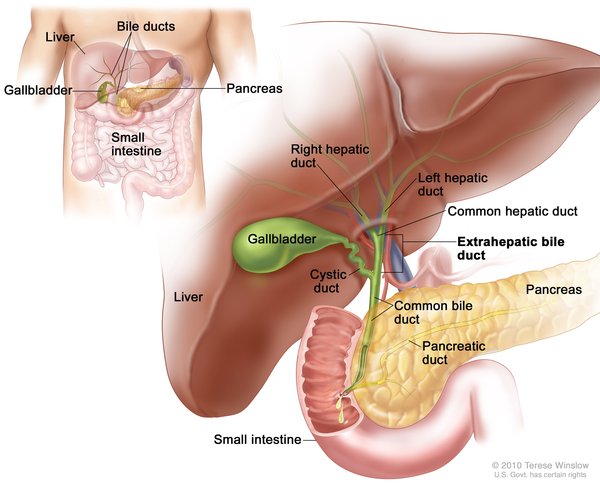
The three types of cholangiocarcinoma do not usually cause any symptoms in their early stages, and this cancer is usually not diagnosed until it has already spread beyond the bile ducts to other tissues. Symptoms often result when bile ducts become blocked by the tumor. The most common symptom is jaundice, in which the skin and whites of the eyes turn yellow. Other symptoms can include extreme tiredness (fatigue), itching, dark-colored urine, loss of appetite, unintentional weight loss, abdominal pain, and light-colored and greasy stools. These symptoms are described as "nonspecific" because they can be features of many different diseases.
Most people who develop cholangiocarcinoma are older than 65. Because this cancer is often not discovered until it has already spread, it can be challenging to treat effectively. Affected individuals can survive for several months to several years after diagnosis, depending on the location of the cancer and how advanced it is.
Frequency
Cholangiocarcinoma affects 8,000 people each year in the United States. This type of cancer occurs much more frequently in Southeast Asian countries such as Thailand, where it is related to infection with a parasite that is common there. For unknown reasons, cholangiocarcinoma occurs slightly more often in men than in women.
Causes
Cancers occur when a buildup of mutations in critical genes—those that control cell division, for example—allow cells to grow and divide uncontrollably to form a tumor. In most cases of cholangiocarcinoma, these genetic changes are acquired during a person's lifetime and are present only in the bile duct cells that give rise to the tumor. The genetic changes, which are called somatic mutations, are not inherited. Somatic mutations in many different genes have been found in cholangiocarcinoma. Some of these genes act as tumor suppressors, which means they help keep the growth and division of cells tightly regulated. Mutations in or deletions of tumor suppressor genes can allow cells to grow and divide without control or order, which is a hallmark of cancer. Other genes associated with cholangiocarcinoma are oncogenes; when they are turned on (activated) abnormally, these genes have the potential to cause normal cells to become cancerous. Identifying somatic mutations in cholangiocarcinoma may provide clues to how quickly the cancer will grow and spread, and which treatments might be most effective.
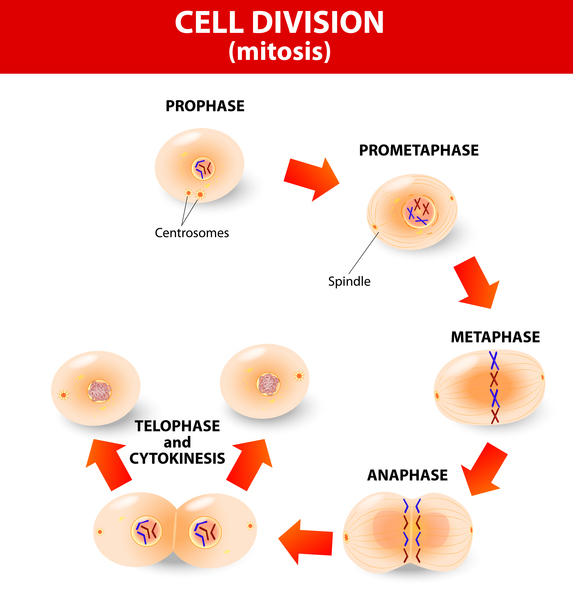
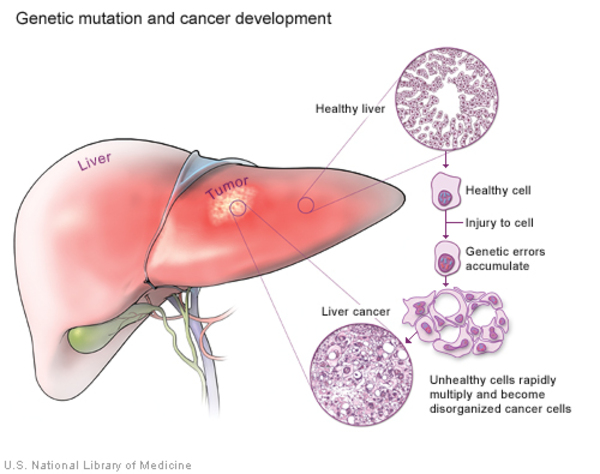
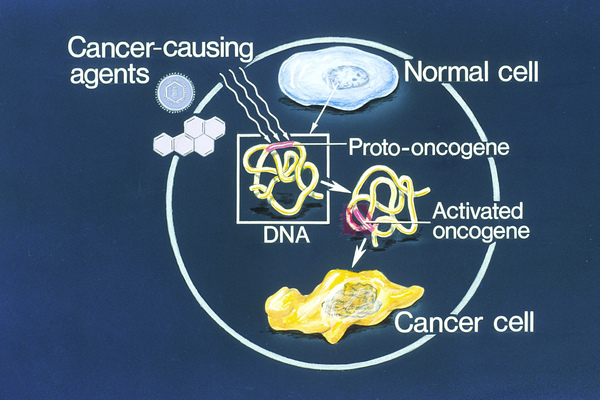
Researchers have also investigated inherited variations in several genes as possible risk factors for cholangiocarcinoma. These genetic changes, which are classified as germline mutations, are present in essentially all of the body's cells. However, no specific inherited changes have been found to be major risk factors for this disease.
Several non-genetic risk factors for cholangiocarcinoma have been identified. These include a bile duct disease called primary sclerosing cholangitis, bile duct stones or cysts, and exposure to certain chemical toxins used in manufacturing. In Southeast Asia, infection with parasitic worms that live in the human bile ducts greatly increase the risk of developing cholangiocarcinoma. Other risk factors that have been studied include long-term infection with viral hepatitis B or C, scarring of the liver (cirrhosis), and chronic diseases such as inflammatory bowel disease and diabetes. Researchers suspect that certain lifestyle factors, including smoking, alcohol use, and obesity, may also contribute to the risk of developing cholangiocarcinoma.
Studies suggest that a combination of genetic, environmental, and lifestyle factors influence whether a person will develop cholangiocarcinoma. However, most people who develop the disease do not have any of the identified risk factors.
Inheritance
Cholangiocarcinoma is not inherited. Studies suggest that blood relatives of a person with cholangiocarcinoma may have an increased risk of developing this cancer compared with the general population. However, most people with cholangiocarcinoma do not have a family history of the disease.
Other Names for This Condition
- CC
- Cholangiocarcinoma of biliary tract
- Cholangiocellular carcinoma
- Distal cholangiocarcinoma
- Extrahepatic cholangiocarcinoma
- Intrahepatic cholangiocarcinoma
- Perihilar cholangiocarcinoma
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Banales JM, Cardinale V, Carpino G, Marzioni M, Andersen JB, Invernizzi P, Lind GE, Folseraas T, Forbes SJ, Fouassier L, Geier A, Calvisi DF, Mertens JC, Trauner M, Benedetti A, Maroni L, Vaquero J, Macias RI, Raggi C, Perugorria MJ, Gaudio E, Boberg KM, Marin JJ, Alvaro D. Expert consensus document: Cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol. 2016 May;13(5):261-80. doi: 10.1038/nrgastro.2016.51. Epub 2016 Apr 20. Citation on PubMed
- Bergquist A, von Seth E. Epidemiology of cholangiocarcinoma. Best Pract Res Clin Gastroenterol. 2015 Apr;29(2):221-32. doi: 10.1016/j.bpg.2015.02.003. Epub 2015 Feb 16. Citation on PubMed
- Kongpetch S, Jusakul A, Ong CK, Lim WK, Rozen SG, Tan P, Teh BT. Pathogenesis of cholangiocarcinoma: From genetics to signalling pathways. Best Pract Res Clin Gastroenterol. 2015 Apr;29(2):233-44. doi: 10.1016/j.bpg.2015.02.002. Epub 2015 Feb 17. Citation on PubMed
- Maroni L, Pierantonelli I, Banales JM, Benedetti A, Marzioni M. The significance of genetics for cholangiocarcinoma development. Ann Transl Med. 2013 Oct;1(3):28. doi: 10.3978/j.issn.2305-5839.2012.10.04. Citation on PubMed or Free article on PubMed Central
- Plentz RR, Malek NP. Clinical presentation, risk factors and staging systems of cholangiocarcinoma. Best Pract Res Clin Gastroenterol. 2015 Apr;29(2):245-52. doi: 10.1016/j.bpg.2015.02.001. Epub 2015 Feb 14. Citation on PubMed
- Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014 Jun 21;383(9935):2168-79. doi: 10.1016/S0140-6736(13)61903-0. Epub 2014 Feb 26. Citation on PubMed or Free article on PubMed Central
- Valle JW, Lamarca A, Goyal L, Barriuso J, Zhu AX. New Horizons for Precision Medicine in Biliary Tract Cancers. Cancer Discov. 2017 Sep;7(9):943-962. doi: 10.1158/2159-8290.CD-17-0245. Epub 2017 Aug 17. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.