

Description
Chediak-Higashi syndrome is a condition that affects many parts of the body, particularly the immune system. This disease damages immune system cells, leaving them less able to fight off invaders such as viruses and bacteria. As a result, most people with Chediak-Higashi syndrome have repeated and persistent infections starting in infancy or early childhood. These infections tend to be very serious or life-threatening.
Chediak-Higashi syndrome is also characterized by a condition called oculocutaneous albinism, which causes abnormally light coloring (pigmentation) of the skin, hair, and eyes. Affected individuals typically have fair skin and light-colored hair, often with a metallic sheen. Oculocutaneous albinism also causes vision problems such as reduced sharpness; rapid, involuntary eye movements (nystagmus); and increased sensitivity to light (photophobia).

Many people with Chediak-Higashi syndrome have problems with blood clotting (coagulation) that lead to easy bruising and abnormal bleeding. In adulthood, Chediak-Higashi syndrome can also affect the nervous system, causing weakness, clumsiness, difficulty with walking, and seizures.
If the disease is not successfully treated, most children with Chediak-Higashi syndrome reach a stage of the disorder known as the accelerated phase. This severe phase of the disease is thought to be triggered by a viral infection. In the accelerated phase, white blood cells (which normally help fight infection) divide uncontrollably and invade many of the body's organs. The accelerated phase is associated with fever, episodes of abnormal bleeding, overwhelming infections, and organ failure. These medical problems are usually life-threatening in childhood.
A small percentage of people with Chediak-Higashi syndrome have a milder form of the condition that appears later in life. People with the adult form of the disorder have less noticeable changes in pigmentation and are less likely to have recurrent, severe infections. They do, however, have a significant risk of progressive neurological problems such as tremors, difficulty with movement and balance (ataxia), reduced sensation and weakness in the arms and legs (peripheral neuropathy), and a decline in intellectual functioning.
Frequency
Chediak-Higashi syndrome is a rare disorder. About 200 cases of the condition have been reported worldwide.
Causes
Chediak-Higashi syndrome is caused by mutations in the LYST gene. This gene provides instructions for making a protein known as the lysosomal trafficking regulator. Researchers believe that this protein plays a role in the transport (trafficking) of materials into structures called lysosomes and similar cell structures. Lysosomes act as recycling centers within cells. They use digestive enzymes to break down toxic substances, digest bacteria that invade the cell, and recycle worn-out cell components.
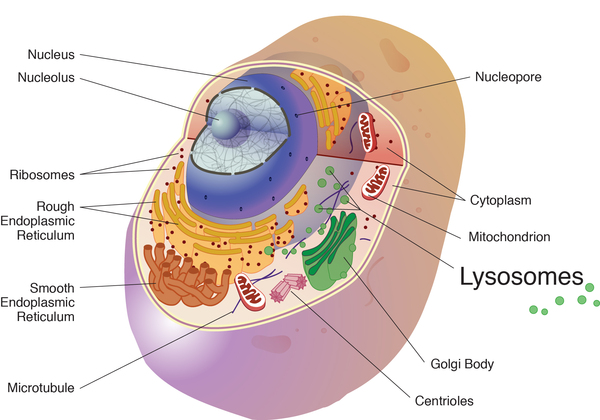
Mutations in the LYST gene impair the normal function of the lysosomal trafficking regulator protein, which disrupts the size, structure, and function of lysosomes and related structures in cells throughout the body. In many cells, the lysosomes are abnormally large and interfere with normal cell functions. For example, enlarged lysosomes in certain immune system cells prevent these cells from responding appropriately to bacteria and other foreign invaders. As a result, the malfunctioning immune system cannot protect the body from infections.
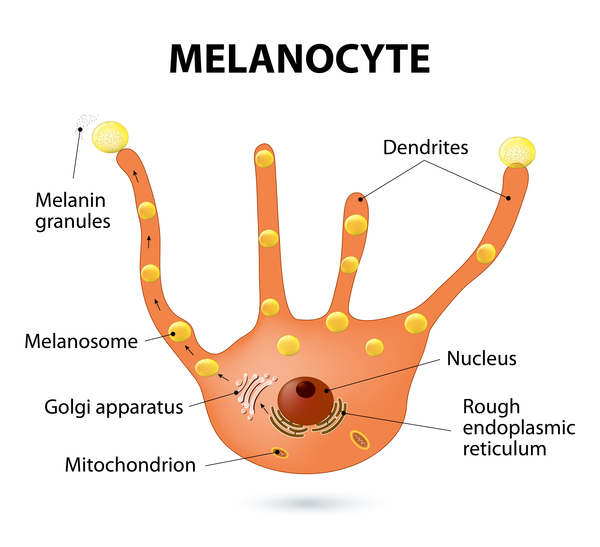
In pigment cells called melanocytes, cellular structures called melanosomes (which are related to lysosomes) are abnormally large. Melanosomes produce and distribute a pigment called melanin, which is the substance that gives skin, hair, and eyes their color. People with Chediak-Higashi syndrome have oculocutaneous albinism because melanin is trapped within the giant melanosomes and is unable to contribute to skin, hair, and eye pigmentation.
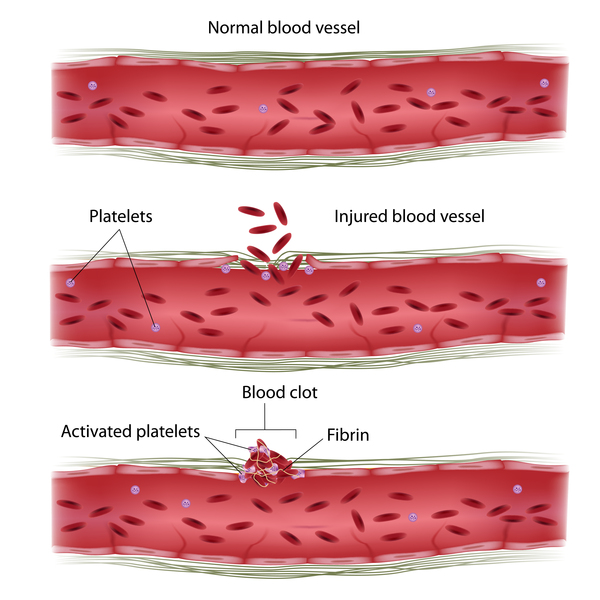
Researchers believe that abnormal lysosome-like structures inside blood cells called platelets underlie the abnormal bruising and bleeding seen in people with Chediak-Higashi syndrome. Similarly, abnormal lysosomes in nerve cells probably cause the neurological problems associated with this disease.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Chediak-Steinbrinck-Higashi syndrome
- CHS
- Oculocutaneous albinism with leukocyte defect
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Huizing M, Anikster Y, Gahl WA. Hermansky-Pudlak syndrome and Chediak-Higashi syndrome: disorders of vesicle formation and trafficking. Thromb Haemost. 2001 Jul;86(1):233-45. Citation on PubMed
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol Genet Metab. 1999 Oct;68(2):283-303. doi: 10.1006/mgme.1999.2927. Citation on PubMed
- Kaplan J, De Domenico I, Ward DM. Chediak-Higashi syndrome. Curr Opin Hematol. 2008 Jan;15(1):22-9. doi: 10.1097/MOH.0b013e3282f2bcce. Citation on PubMed
- Karim MA, Suzuki K, Fukai K, Oh J, Nagle DL, Moore KJ, Barbosa E, Falik-Borenstein T, Filipovich A, Ishida Y, Kivrikko S, Klein C, Kreuz F, Levin A, Miyajima H, Regueiro JR, Russo C, Uyama E, Vierimaa O, Spritz RA. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak-Higashi syndrome. Am J Med Genet. 2002 Feb 15;108(1):16-22. Citation on PubMed
- Ward DM, Shiflett SL, Kaplan J. Chediak-Higashi syndrome: a clinical and molecular view of a rare lysosomal storage disorder. Curr Mol Med. 2002 Aug;2(5):469-77. doi: 10.2174/1566524023362339. Citation on PubMed
- Westbroek W, Adams D, Huizing M, Koshoffer A, Dorward H, Tinloy B, Parkes J, Helip-Wooley A, Kleta R, Tsilou E, Duvernay P, Digre KB, Creel DJ, White JG, Boissy RE, Gahl WA. Cellular defects in Chediak-Higashi syndrome correlate with the molecular genotype and clinical phenotype. J Invest Dermatol. 2007 Nov;127(11):2674-7. doi: 10.1038/sj.jid.5700899. Epub 2007 May 31. No abstract available. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.