

Description
Carpenter syndrome is a condition characterized by the premature fusion of certain skull bones (craniosynostosis), abnormalities of the fingers and toes, and other developmental problems.
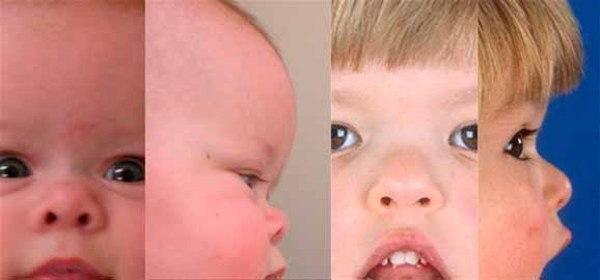
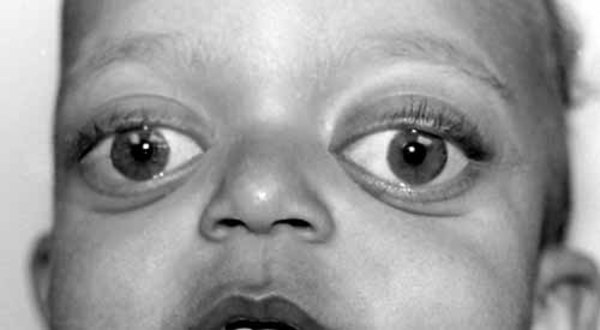
Craniosynostosis prevents the skull from growing normally, frequently giving the head a pointed appearance (acrocephaly). In severely affected individuals, the abnormal fusion of the skull bones results in a deformity called a cloverleaf skull. Craniosynostosis can cause differences between the two sides of the head and face (craniofacial asymmetry). Early fusion of the skull bones can affect the development of the brain and lead to increased pressure within the skull (intracranial pressure). Premature fusion of the skull bones can cause several characteristic facial features in people with Carpenter syndrome. Distinctive facial features may include a flat nasal bridge, outside corners of the eyes that point downward (down-slanting palpebral fissures), low-set and abnormally shaped ears, underdeveloped upper and lower jaws, and abnormal eye shape. Some affected individuals also have dental abnormalities including small primary (baby) teeth. Vision problems also frequently occur.
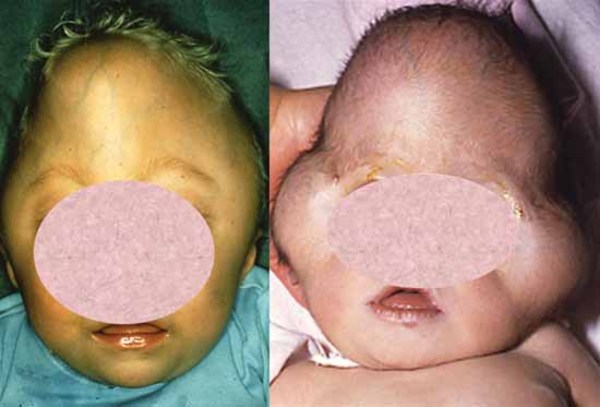
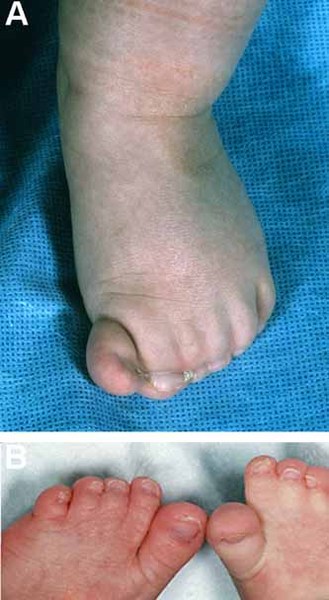
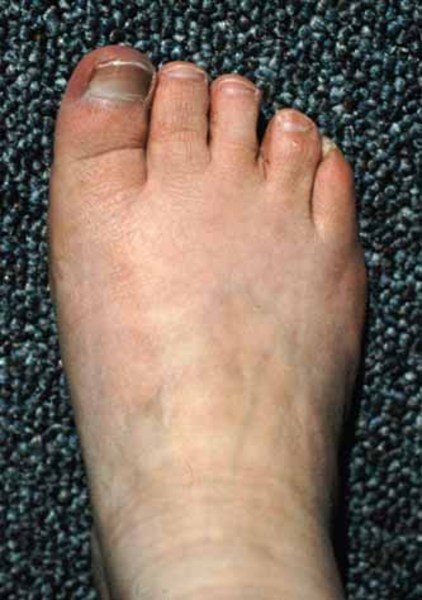
Abnormalities of the fingers and toes include fusion of the skin between two or more fingers or toes (cutaneous syndactyly), unusually short fingers or toes (brachydactyly), or extra fingers or toes (polydactyly). In Carpenter syndrome, cutaneous syndactyly is most common between the third (middle) and fourth (ring) fingers, and polydactyly frequently occurs next to the big or second toe or the fifth (pinky) finger.
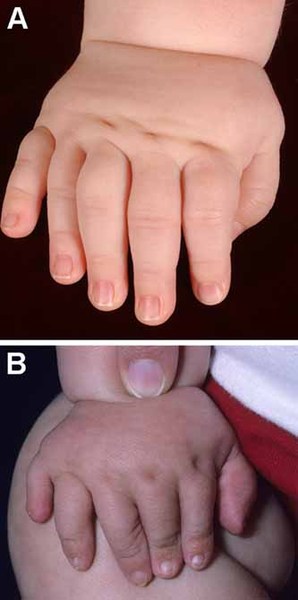
People with Carpenter syndrome often have intellectual disability, which can range from mild to profound. However, some individuals with this condition have normal intelligence. The cause of intellectual disability is unknown, as the severity of craniosynostosis does not appear to be related to the severity of intellectual disability.
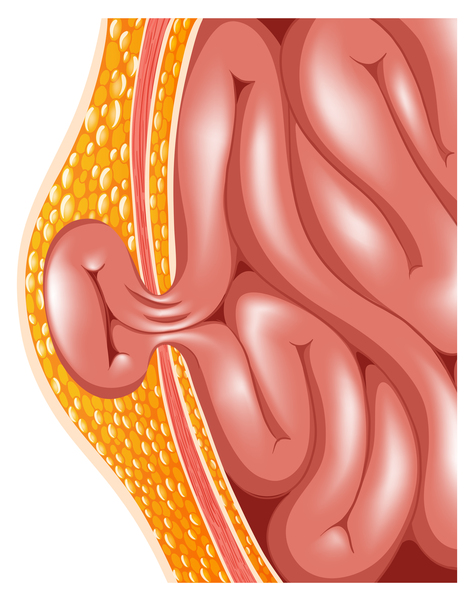
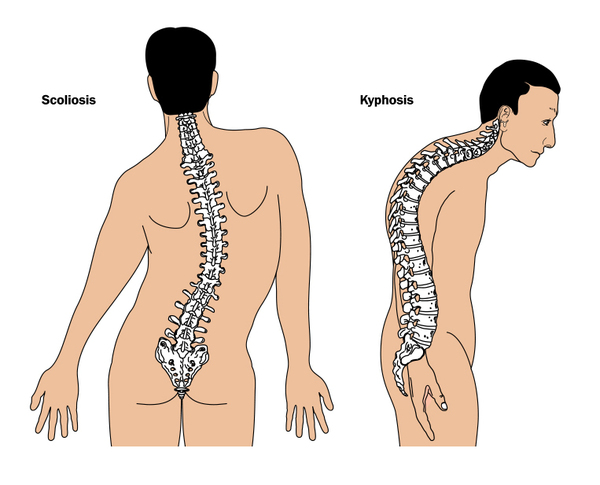
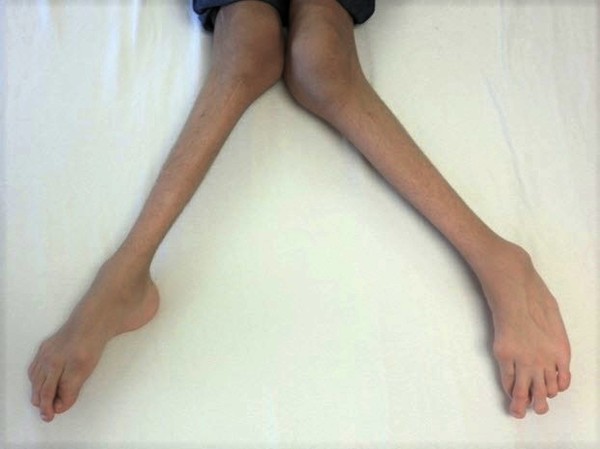
Other features of Carpenter syndrome include obesity that begins in childhood, a soft out-pouching around the belly-button (umbilical hernia), hearing loss, and heart defects. Additional skeletal abnormalities such as deformed hips, a rounded upper back that also curves to the side (kyphoscoliosis), and knees that are angled inward (genu valgum) frequently occur. Nearly all affected males have genital abnormalities, most frequently undescended testes (cryptorchidism).
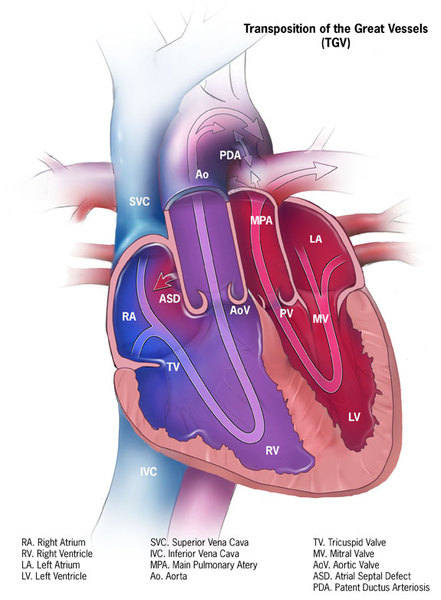
A few people with Carpenter syndrome have organs or tissues within their chest and abdomen that are in mirror-image reversed positions. This abnormal placement may affect several internal organs (situs inversus); just the heart (dextrocardia), placing the heart on the right side of the body instead of on the left; or only the major (great) arteries of the heart, altering blood flow.
The signs and symptoms of this disorder vary considerably, even within the same family. The life expectancy for individuals with Carpenter syndrome is shortened but extremely variable.
The signs and symptoms of Carpenter syndrome are similar to another genetic condition called Greig cephalopolysyndactyly syndrome. The overlapping features, which include craniosynostosis, polydactyly, and heart abnormalities, can cause these two conditions to be misdiagnosed; genetic testing is often required for an accurate diagnosis.
Frequency
Carpenter syndrome is thought to be a rare condition; approximately 70 cases have been described in the scientific literature.
Causes
Mutations in the RAB23 or MEGF8 gene cause Carpenter syndrome.
The RAB23 gene provides instructions for making a protein that is involved in a process called vesicle trafficking, which moves proteins and other molecules within cells in sac-like structures called vesicles. The Rab23 protein transports vesicles from the cell membrane to their proper location inside the cell. Vesicle trafficking is important for the transport of materials that are needed to trigger signaling during development. For example, the Rab23 protein regulates a developmental pathway called the hedgehog signaling pathway that is critical in cell growth (proliferation), cell specialization, and the normal shaping (patterning) of many parts of the body.

The MEGF8 gene provides instructions for making a protein whose function is unclear. Based on its structure, the Megf8 protein may be involved in cell processes such as sticking cells together (cell adhesion) and helping proteins interact with each other. Researchers also suspect that the Megf8 protein plays a role in normal body patterning.
Mutations in the RAB23 or MEGF8 gene lead to the production of proteins with little or no function. It is unclear how disruptions in protein function lead to the features of Carpenter syndrome, but it is likely that interference with normal body patterning plays a role. For reasons that are unknown, people with MEGF8 gene mutations are more likely to have dextrocardia and other organ positioning abnormalities and less severe craniosynostosis than individuals with RAB23 gene mutations.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- ACPS II
- Acrocephalopolysyndactyly 2
- Acrocephalopolysyndactyly type II
- Acrocephalosyndactyly, type II
- Type II acrocephalosyndactyly
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Alessandri JL, Dagoneau N, Laville JM, Baruteau J, Hebert JC, Cormier-Daire V. RAB23 mutation in a large family from Comoros Islands with Carpenter syndrome. Am J Med Genet A. 2010 Apr;152A(4):982-6. doi: 10.1002/ajmg.a.33327. Citation on PubMed
- Hidestrand P, Vasconez H, Cottrill C. Carpenter syndrome. J Craniofac Surg. 2009 Jan;20(1):254-6. doi: 10.1097/SCS.0b013e318184357a. Citation on PubMed
- Jenkins D, Baynam G, De Catte L, Elcioglu N, Gabbett MT, Hudgins L, Hurst JA, Jehee FS, Oley C, Wilkie AO. Carpenter syndrome: extended RAB23 mutation spectrum and analysis of nonsense-mediated mRNA decay. Hum Mutat. 2011 Apr;32(4):E2069-78. doi: 10.1002/humu.21457. Epub 2011 Feb 8. Citation on PubMed or Free article on PubMed Central
- Jenkins D, Seelow D, Jehee FS, Perlyn CA, Alonso LG, Bueno DF, Donnai D, Josifova D, Mathijssen IM, Morton JE, Orstavik KH, Sweeney E, Wall SA, Marsh JL, Nurnberg P, Passos-Bueno MR, Wilkie AO. RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am J Hum Genet. 2007 Jun;80(6):1162-70. doi: 10.1086/518047. Epub 2007 Apr 18. Citation on PubMed or Free article on PubMed Central
- Perlyn CA, Marsh JL. Craniofacial dysmorphology of Carpenter syndrome: lessons from three affected siblings. Plast Reconstr Surg. 2008 Mar;121(3):971-981. doi: 10.1097/01.prs.0000299284.92862.6c. Citation on PubMed
- Ramos JM, Davis GJ, Hunsaker JC 3rd, Balko MG. Sudden death in a child with Carpenter Syndrome. Case report and literature review. Forensic Sci Med Pathol. 2009 Dec;5(4):313-7. doi: 10.1007/s12024-009-9128-2. Epub 2009 Nov 19. Citation on PubMed
- Twigg SR, Lloyd D, Jenkins D, Elcioglu NE, Cooper CD, Al-Sannaa N, Annagur A, Gillessen-Kaesbach G, Huning I, Knight SJ, Goodship JA, Keavney BD, Beales PL, Gileadi O, McGowan SJ, Wilkie AO. Mutations in multidomain protein MEGF8 identify a Carpenter syndrome subtype associated with defective lateralization. Am J Hum Genet. 2012 Nov 2;91(5):897-905. doi: 10.1016/j.ajhg.2012.08.027. Epub 2012 Oct 11. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.