

Description
Bohring-Opitz syndrome is a rare condition that affects the development of many parts of the body.
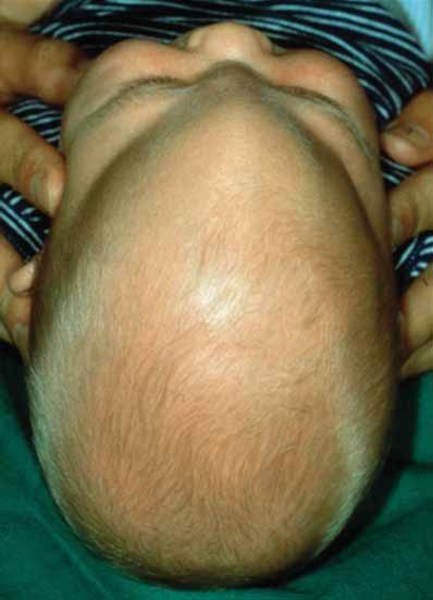
Most individuals with Bohring-Opitz syndrome have profound to severe intellectual disability, developmental delay, and seizures. Most affected individuals have a normal head shape and size with no brain abnormalities; however, some have abnormal development of the head. Abnormal development can lead to a small head size (microcephaly) and a skull abnormality called trigonocephaly, which gives the forehead a pointed appearance. Structural brain abnormalities can occur with or without head abnormalities. For example, the fluid-filled spaces near the center of the brain (ventricles) may be usually large (ventriculomegaly) or the tissue that connects the left and right halves of the brain (the corpus callosum) can be abnormally thin.

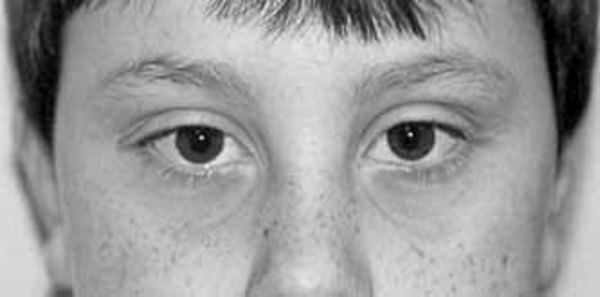
Eye problems that can affect vision also occur in people with Bohring-Opitz syndrome. People with this disorder may have protruding eyes (exophthalmos), eyes that do not point in the same direction (strabismus), widely spaced eyes (hypertelorism), or outside corners of the eyes that point upward (upslanting palpebral fissures). Affected individuals may have severe nearsightedness (high myopia) or abnormalities in the light-sensitive tissue at the back of the eye (the retina) or the nerves that carry information from the eyes to the brain (optic nerves).

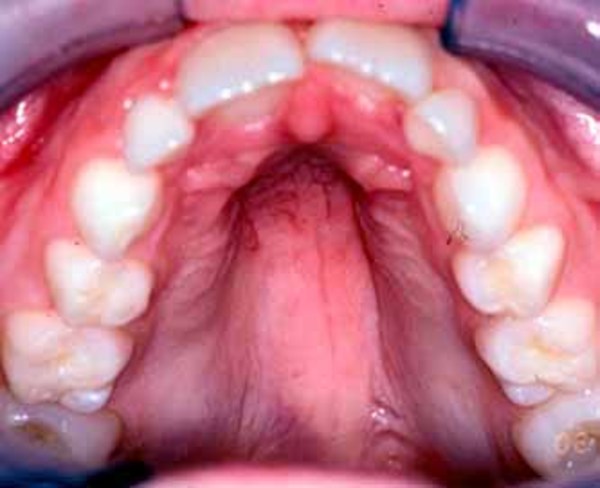
Additional facial differences associated with Bohring-Opitz syndrome can include a flat nasal bridge, nostrils that open to the front rather than downward (anteverted nares), a high arch or opening in the roof of the mouth (high arched or cleft palate), a split in the upper lip (cleft lip), a small lower jaw (micrognathia), low-set ears that are rotated backward, a red birthmark (nevus simplex) on the face (usually the forehead), a low frontal hairline often with eyebrows that grow together in the middle (synophrys), and excessive body and facial hair (hirsutism) that increases with age.
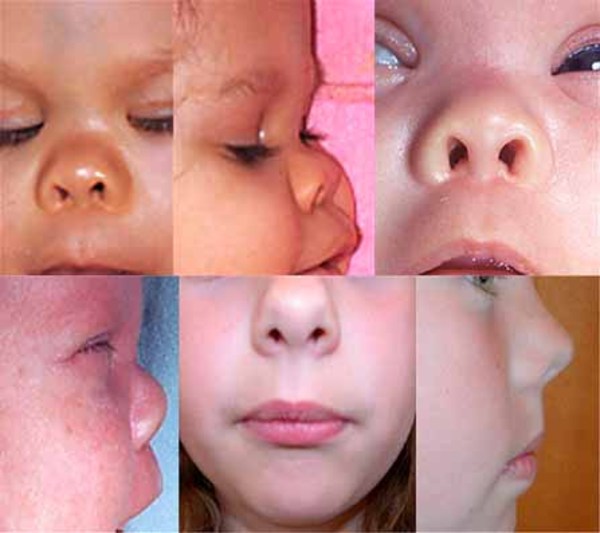

Some individuals with Bohring-Opitz syndrome have poor growth before birth (intrauterine growth retardation). During infancy, they grow and gain weight slowly and often have severe feeding difficulties with recurrent vomiting.
People with this condition often have characteristic body positioning, known as Bohring-Opitz syndrome posture. This posture consists of slouching shoulders, bent elbows and wrists, hands positioned with the wrists or all of the fingers angled outward toward the fifth finger (ulnar deviation), with the legs usually extended straight. Affected individuals usually stop exhibiting the Bohring-Opitz syndrome posture as they get older. Other abnormalities include joint deformities (called contractures) that are apparent at birth in the knees, hips, or other joints and abnormal muscle tone. Affected individuals can have recurrent infections and heart, kidney, or genital abnormalities. In rare cases, a childhood form of kidney cancer known as Wilms tumor can develop.
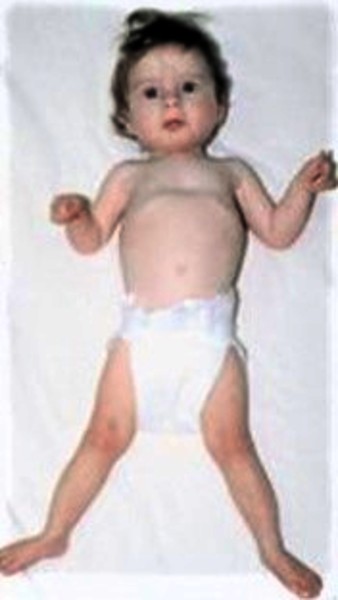

Some individuals with Bohring-Opitz syndrome do not survive past early childhood, while others live into adolescence or early adulthood. The most common causes of death are heart problems, abnormalities of the throat and airways that cause pauses in breathing (obstructive apnea), and lung infections.
Frequency
Bohring-Opitz syndrome is thought to be a rare condition, although its exact prevalence is unknown. More than 40 affected individuals have been described in the scientific literature.
Causes
Bohring-Opitz syndrome is caused by mutations in the ASXL1 gene. This gene provides instructions for making a protein that is involved in a process known as chromatin remodeling. Chromatin is the complex of DNA and proteins that packages DNA into chromosomes. The structure of chromatin can be changed (remodeled) to alter how tightly DNA is packaged. Through its role in chromatin remodeling, the ASXL1 gene regulates the activity (expression) of many genes, including a group of genes known as HOX genes, which play important roles in development before birth. The ASXL1 protein can turn on (activate) or turn off (repress) HOX genes depending on when they are needed.
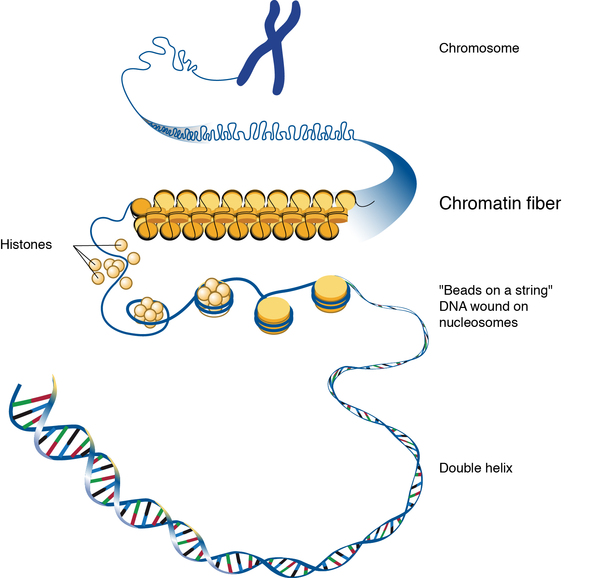
ASXL1 gene mutations reduce the amount of functional ASXL1 protein available, which likely disrupts the regulation of the activity of HOX genes and other genes during development. Altered activity of these genes probably leads to the neurological and physical features of this condition.
Inheritance
Bohring-Opitz syndrome is considered an autosomal dominant condition, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Most cases of the condition result from new (de novo) mutations in the gene that occur during the formation of reproductive cells (eggs or sperm) or in early embryonic development. These cases occur in people with no history of the disorder in their family. No one with Bohring-Opitz syndrome has been known to have children.
Very rarely, individuals with Bohring-Opitz syndrome inherit the altered gene from their unaffected mother, who has the mutation only in some cells, including egg cells, but not in others. This phenomenon is known as mosaicism.
Other Names for This Condition
- Bohring syndrome
- BOPS
- BOS
- C-like syndrome
- Oberklaid-Danks syndrome
- Opitz trigonocephaly-like syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bedoukian E, Copenheaver D, Bale S, Deardorff M. Bohring-Opitz syndrome caused by an ASXL1 mutation inherited from a germline mosaic mother. Am J Med Genet A. 2018 May;176(5):1249-1252. doi: 10.1002/ajmg.a.38686. Citation on PubMed
- Bohring A, Oudesluijs GG, Grange DK, Zampino G, Thierry P. New cases of Bohring-Opitz syndrome, update, and critical review of the literature. Am J Med Genet A. 2006 Jun 15;140(12):1257-63. doi: 10.1002/ajmg.a.31265. Citation on PubMed
- Hoischen A, van Bon BW, Rodriguez-Santiago B, Gilissen C, Vissers LE, de Vries P, Janssen I, van Lier B, Hastings R, Smithson SF, Newbury-Ecob R, Kjaergaard S, Goodship J, McGowan R, Bartholdi D, Rauch A, Peippo M, Cobben JM, Wieczorek D, Gillessen-Kaesbach G, Veltman JA, Brunner HG, de Vries BB. De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat Genet. 2011 Jun 26;43(8):729-31. doi: 10.1038/ng.868. Citation on PubMed
- Magini P, Della Monica M, Uzielli ML, Mongelli P, Scarselli G, Gambineri E, Scarano G, Seri M. Two novel patients with Bohring-Opitz syndrome caused by de novo ASXL1 mutations. Am J Med Genet A. 2012 Apr;158A(4):917-21. doi: 10.1002/ajmg.a.35265. Epub 2012 Mar 14. Citation on PubMed
- Russell B, Johnston JJ, Biesecker LG, Kramer N, Pickart A, Rhead W, Tan WH, Brownstein CA, Kate Clarkson L, Dobson A, Rosenberg AZ, Vergano SA, Helm BM, Harrison RE, Graham JM Jr. Clinical management of patients with ASXL1 mutations and Bohring-Opitz syndrome, emphasizing the need for Wilms tumor surveillance. Am J Med Genet A. 2015 Sep;167A(9):2122-31. doi: 10.1002/ajmg.a.37131. Epub 2015 Apr 29. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.