

Description
Behçet disease is an inflammatory condition that affects many parts of the body. The health problems associated with Behçet disease result from widespread inflammation of blood vessels (vasculitis). This inflammation most commonly affects small blood vessels in the mouth, genitals, skin, and eyes.
Painful mouth sores called aphthous ulcers are usually the first sign of Behçet disease. These sores can occur on the lips, tongue, inside the cheeks, the roof of the mouth, the throat, and the tonsils. The ulcers look like common canker sores, and they typically heal within one to two weeks. About 75 percent of all people with Behçet disease develop similar ulcers on the genitals. These ulcers occur most frequently on the scrotum in men and on the labia in women.
Behçet disease can also cause painful bumps and sores on the skin. Most affected individuals develop pus-filled bumps that resemble acne. These bumps can occur anywhere on the body. Some affected people also have red, tender nodules called erythema nodosum. These nodules usually develop on the legs but can also occur on the arms, face, and neck.
An inflammation of the eye called uveitis is found in more than half of people with Behçet disease. Eye problems are more common in younger people with the disease and affect men more often than women. Uveitis can result in blurry vision and an extreme sensitivity to light (photophobia). Rarely, inflammation can also cause eye pain and redness. If untreated, the eye problems associated with Behçet disease can lead to blindness.
Joint involvement is also common in Behçet disease. Often this affects one joint at a time, with each affected joint becoming swollen and painful and then getting better.
Less commonly, Behçet disease can affect the brain and spinal cord (central nervous system), gastrointestinal tract, large blood vessels, heart, lungs, and kidneys. Central nervous system abnormalities can lead to headaches, confusion, personality changes, memory loss, impaired speech, and problems with balance and movement. Involvement of the gastrointestinal tract can lead to a hole in the wall of the intestine (intestinal perforation), which can cause serious infection and may be life-threatening.
The signs and symptoms of Behçet disease usually begin in a person's twenties or thirties, although they can appear at any age. Some affected people have relatively mild symptoms that are limited to sores in the mouth and on the genitals. Others have more severe symptoms affecting various parts of the body, including the eyes and the vital organs. The features of Behçet disease typically come and go over a period of months or years. In most affected individuals, the health problems associated with this disorder improve with age.
Frequency
Behçet disease is most common in Mediterranean countries, the Middle East, Japan, and other parts of Asia. However, it has been found in populations worldwide.
The highest prevalence of Behçet disease has been reported in northern Turkey, where the disorder affects up to 420 in 100,000 people. The disorder is rare in northern European countries and the United States, where it generally affects fewer than 1 in 100,000 people.
Causes
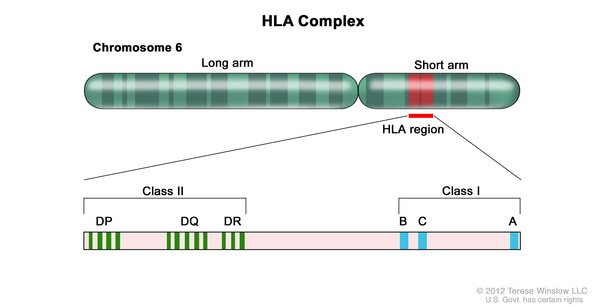
The cause of Behçet disease is unknown. The condition probably results from a combination of genetic and environmental factors, most of which have not been identified. However, a particular variation in the HLA-B gene has been associated with the risk of developing Behçet disease.
The HLA-B gene provides instructions for making a protein that plays an important role in the immune system. The HLA-B gene is part of a family of genes called the human leukocyte antigen (HLA) complex. The HLA complex helps the immune system distinguish the body's own proteins from proteins made by foreign invaders (such as viruses and bacteria). The HLA-B gene has many different normal variations, allowing each person's immune system to react to a wide range of foreign proteins. A variation of the HLA-B gene called HLA-B51 increases the risk of developing Behçet disease by about a factor of six, although the mechanism is not well understood. One-third to two-thirds of people with Behçet disease have the HLA-B51 variation, but most people with this version of the HLA-B gene never develop the disorder.
Other genetic and environmental factors likely contribute to the risk of Behçet disease. Researchers are studying several genes related to immune system function. It also appears likely that environmental factors, such as certain bacterial or viral infections, play a role in triggering the disease in people who are at risk. However, the influence of genetic and environmental factors on the development of this complex disorder remains unclear.
Inheritance
Most cases of Behçet disease are sporadic, which means they occur in people with no history of the disorder in their family. A small percentage of all cases have been reported to run in families; however, the condition does not have a clear pattern of inheritance.
Other Names for This Condition
- Adamantiades-Behcet disease
- Behcet disease
- Behcet syndrome
- Behcet triple symptom complex
- Behcet's syndrome
- Malignant aphthosis
- Old Silk Route disease
- Triple symptom complex
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- de Menthon M, Lavalley MP, Maldini C, Guillevin L, Mahr A. HLA-B51/B5 and the risk of Behcet's disease: a systematic review and meta-analysis of case-control genetic association studies. Arthritis Rheum. 2009 Oct 15;61(10):1287-96. doi: 10.1002/art.24642. Citation on PubMed or Free article on PubMed Central
- Durrani K, Papaliodis GN. The genetics of Adamantiades-Behcet's disease. Semin Ophthalmol. 2008 Jan-Feb;23(1):73-9. doi: 10.1080/08820530701745264. Citation on PubMed
- International Team for the Revision of the International Criteria for Behcet's Disease (ITR-ICBD). The International Criteria for Behcet's Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014 Mar;28(3):338-47. doi: 10.1111/jdv.12107. Epub 2013 Feb 26. Citation on PubMed
- Johns Hopkins Vasculitis Center
- Kaneko F, Togashi A, Saito S, Sakuma H, Oyama N, Nakamura K, Yokota K, Oguma K. Behcet's disease (Adamantiades-Behcet's disease). Clin Dev Immunol. 2011;2011:681956. doi: 10.1155/2011/681956. Epub 2010 Nov 1. Citation on PubMed or Free article on PubMed Central
- Maldini C, Lavalley MP, Cheminant M, de Menthon M, Mahr A. Relationships of HLA-B51 or B5 genotype with Behcet's disease clinical characteristics: systematic review and meta-analyses of observational studies. Rheumatology (Oxford). 2012 May;51(5):887-900. doi: 10.1093/rheumatology/ker428. Epub 2012 Jan 11. Citation on PubMed
- Meguro A, Inoko H, Ota M, Katsuyama Y, Oka A, Okada E, Yamakawa R, Yuasa T, Fujioka T, Ohno S, Bahram S, Mizuki N. Genetics of Behcet disease inside and outside the MHC. Ann Rheum Dis. 2010 Apr;69(4):747-54. doi: 10.1136/ard.2009.108571. Epub 2009 Aug 13. Citation on PubMed
- Mendes D, Correia M, Barbedo M, Vaio T, Mota M, Goncalves O, Valente J. Behcet's disease--a contemporary review. J Autoimmun. 2009 May-Jun;32(3-4):178-88. doi: 10.1016/j.jaut.2009.02.011. Epub 2009 Mar 26. Citation on PubMed
- Yazici H, Fresko I, Yurdakul S. Behcet's syndrome: disease manifestations, management, and advances in treatment. Nat Clin Pract Rheumatol. 2007 Mar;3(3):148-55. doi: 10.1038/ncprheum0436. Citation on PubMed
- Yurdakul S, Yazici H. Behcet's syndrome. Best Pract Res Clin Rheumatol. 2008 Oct;22(5):793-809. doi: 10.1016/j.berh.2008.08.005. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.