

Description
Baller-Gerold syndrome is a rare condition characterized by the premature fusion of certain skull bones (craniosynostosis) and abnormalities of bones in the arms and hands.
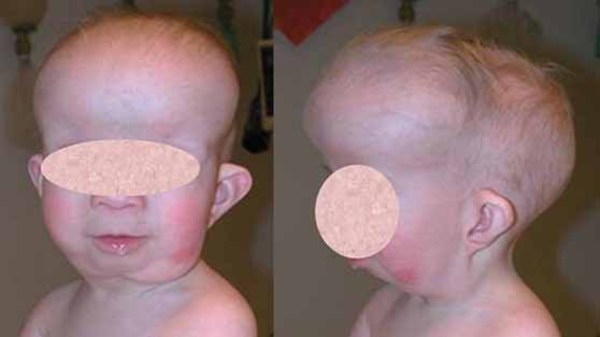
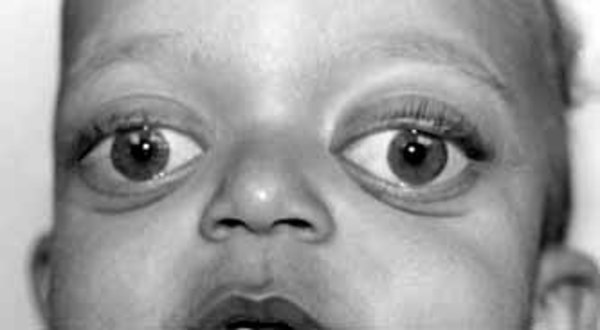
People with Baller-Gerold syndrome have prematurely fused skull bones, most often along the coronal suture, the growth line that goes over the head from ear to ear. Other sutures of the skull may be fused as well. These changes result in an abnormally shaped head, a prominent forehead, and bulging eyes with shallow eye sockets (ocular proptosis). Other distinctive facial features can include widely spaced eyes (hypertelorism), a small mouth, and a saddle-shaped or underdeveloped nose.


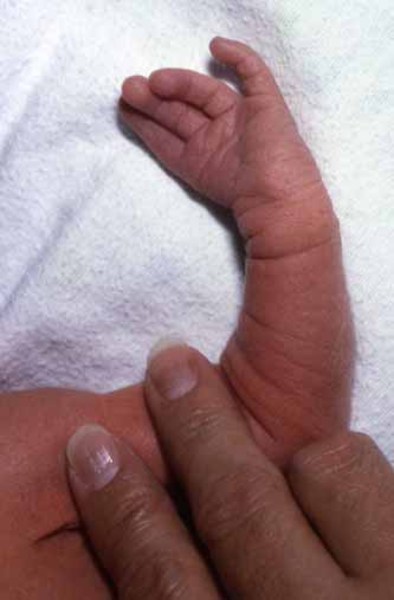
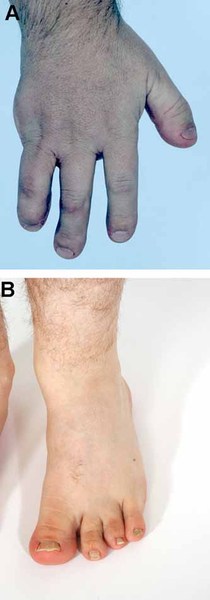
Bone abnormalities in the hands include missing fingers (oligodactyly) and malformed or absent thumbs. Partial or complete absence of bones in the forearm is also common. Together, these hand and arm abnormalities are called radial ray malformations.

People with Baller-Gerold syndrome may have a variety of additional signs and symptoms including slow growth beginning in infancy, small stature, and malformed or missing kneecaps (patellae). A skin rash often appears on the arms and legs a few months after birth. This rash spreads over time, causing patchy changes in skin coloring, areas of thinning skin (atrophy), and small clusters of blood vessels just under the skin (telangiectases). These chronic skin problems are collectively known as poikiloderma.
The varied signs and symptoms of Baller-Gerold syndrome overlap with features of other disorders, namely Rothmund-Thomson syndrome and RAPADILINO syndrome. These syndromes are also characterized by radial ray defects, skeletal abnormalities, and slow growth. All of these conditions can be caused by mutations in the same gene. Based on these similarities, researchers are investigating whether Baller-Gerold syndrome, Rothmund-Thomson syndrome, and RAPADILINO syndrome are separate disorders or part of a single syndrome with overlapping signs and symptoms.
Frequency
The prevalence of Baller-Gerold syndrome is unknown, but this rare condition probably affects fewer than 1 per million people. Fewer than 40 cases have been reported in the medical literature.
Causes
Mutations in the RECQL4 gene cause some cases of Baller-Gerold syndrome. This gene provides instructions for making one member of a protein family called RecQ helicases. Helicases are enzymes that bind to DNA and temporarily unwind the two spiral strands (double helix) of the DNA molecule. This unwinding is necessary for copying (replicating) DNA in preparation for cell division, and for repairing damaged DNA. The RECQL4 protein helps stabilize genetic information in the body's cells and plays a role in replicating and repairing DNA.
Mutations in the RECQL4 gene prevent cells from producing any RECQL4 protein or change the way the protein is pieced together, which disrupts its usual function. A shortage of this protein may prevent normal DNA replication and repair, causing widespread damage to a person's genetic information over time. It is unclear how a loss of this protein's activity leads to the signs and symptoms of Baller-Gerold syndrome.
This condition has been associated with prenatal (before birth) exposure to a drug called sodium valproate. This medication is used to treat epilepsy and certain psychiatric disorders. Some infants whose mothers took sodium valproate during pregnancy were born with the characteristic features of Baller-Gerold syndrome, such as an unusual skull shape, distinctive facial features, and abnormalities of the arms and hands. However, it is unclear if exposure to the medication caused the condition.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- BGS
- Craniosynostosis with radial defects
- Craniosynostosis-radial aplasia syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Anyane-Yeboa K, Gunning L, Bloom AD. Baller-Gerold syndrome craniosynostosis-radial aplasia syndrome. Clin Genet. 1980 Feb;17(2):161-6. doi: 10.1111/j.1399-0004.1980.tb00126.x. Citation on PubMed
- Feingold M, Sklower SL, Willner JP, Desnick RH, Cohen MM. Craniosynostosis-radial aplasia: Baller-Gerold syndrome. Am J Dis Child. 1979 Dec;133(12):1279-80. doi: 10.1001/archpedi.1979.02130120071014. No abstract available. Citation on PubMed
- Santos de Oliveira R, Lajeunie E, Arnaud E, Renier D. Fetal exposure to sodium valproate associated with Baller-Gerold syndrome: case report and review of the literature. Childs Nerv Syst. 2006 Jan;22(1):90-4. doi: 10.1007/s00381-004-1089-x. Epub 2005 Mar 23. Citation on PubMed
- Van Maldergem L, Piard J, Larizza L, Wang LL. Baller-Gerold Syndrome. 2007 Aug 13 [updated 2018 Apr 19]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1204/ Citation on PubMed
- Van Maldergem L, Siitonen HA, Jalkh N, Chouery E, De Roy M, Delague V, Muenke M, Jabs EW, Cai J, Wang LL, Plon SE, Fourneau C, Kestila M, Gillerot Y, Megarbane A, Verloes A. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J Med Genet. 2006 Feb;43(2):148-52. doi: 10.1136/jmg.2005.031781. Epub 2005 Jun 17. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.