

Description
RAPADILINO syndrome is a rare condition that involves many parts of the body. Bone development is especially affected, causing many of the characteristic features of the condition.
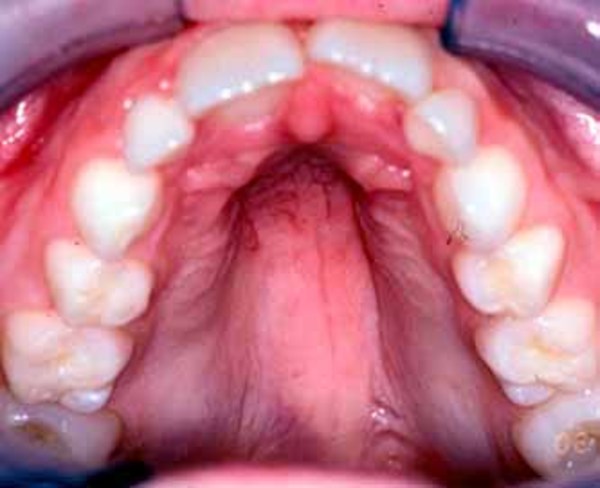
Most affected individuals have underdevelopment or absence of the bones in the forearms and the thumbs, which are known as radial ray malformations. The kneecaps (patellae) can also be underdeveloped or absent. Other features include an opening in the roof of the mouth (cleft palate) or a high arched palate; a long, slender nose; and dislocated joints.
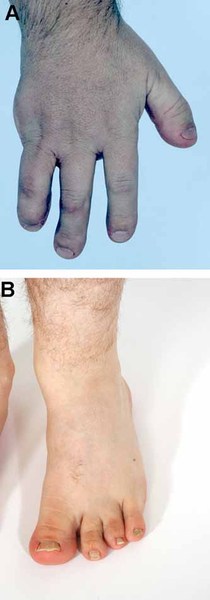

Many infants with RAPADILINO syndrome have difficulty feeding and experience diarrhea and vomiting. The combination of impaired bone development and feeding problems leads to slow growth and short stature in affected individuals.
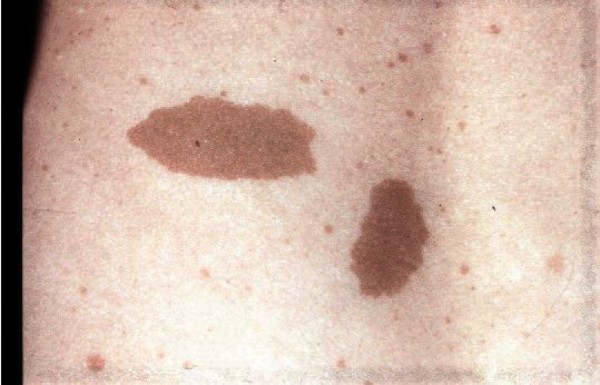
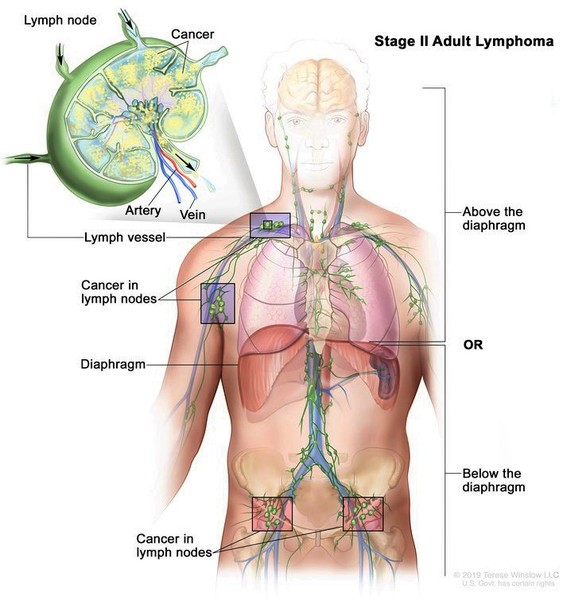
Some individuals with RAPADILINO syndrome have harmless light brown patches of skin that resemble a skin finding known as café-au-lait spots. In addition, people with RAPADILINO syndrome have a slightly increased risk of developing a type of bone cancer known as osteosarcoma or a blood-related cancer called lymphoma. In individuals with RAPADILINO syndrome, osteosarcoma most often develops during childhood or adolescence, and lymphoma typically develops in young adulthood.
The condition name is an acronym for the characteristic features of the disorder: RA for radial ray malformations, PA for patella and palate abnormalities, DI for diarrhea and dislocated joints, LI for limb abnormalities and little size, and NO for slender nose and normal intelligence.
The varied signs and symptoms of RAPADILINO syndrome overlap with features of other disorders, namely Baller-Gerold syndrome and Rothmund-Thomson syndrome. These syndromes are also characterized by radial ray defects, skeletal abnormalities, and slow growth. All of these conditions can be caused by mutations in the same gene. Based on these similarities, researchers are investigating whether Baller-Gerold syndrome, Rothmund-Thomson syndrome, and RAPADILINO syndrome are separate disorders or part of a single syndrome with overlapping signs and symptoms.
Frequency
RAPADILINO syndrome is a rare condition, although its worldwide prevalence is unknown. The condition was first identified in Finland, where it affects an estimated 1 in 75,000 individuals, although it has since been found in other regions.
Causes
Mutations in the RECQL4 gene cause RAPADILINO syndrome. This gene provides instructions for making one member of a protein family called RecQ helicases. Helicases are enzymes that bind to DNA and temporarily unwind the two spiral strands (double helix) of the DNA molecule. This unwinding is necessary for copying (replicating) DNA in preparation for cell division and for repairing damaged DNA. The RECQL4 protein helps stabilize genetic information in the body's cells and plays a role in replicating and repairing DNA.
The most common RECQL4 gene mutation involved in RAPADILINO syndrome causes the RECQL4 protein to be pieced together incorrectly. This genetic change results in the production of a protein that is missing a region called exon 7 and is unable to act as a helicase. The loss of helicase function may prevent normal DNA replication and repair, causing widespread damage to a person's genetic information over time. These changes may result in the accumulation of DNA errors and cell death, although it is unclear exactly how RECQL4 gene mutations lead to the specific features of RAPADILINO syndrome.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Absent thumbs, dislocated joints, long face with narrow palpebral fissures, long slender nose, arched palate
- Radial and patellar aplasia
- Radial and patellar hypoplasia
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Croteau DL, Rossi ML, Ross J, Dawut L, Dunn C, Kulikowicz T, Bohr VA. RAPADILINO RECQL4 mutant protein lacks helicase and ATPase activity. Biochim Biophys Acta. 2012 Nov;1822(11):1727-34. doi: 10.1016/j.bbadis.2012.07.014. Epub 2012 Jul 31. Citation on PubMed or Free article on PubMed Central
- Siitonen HA, Kopra O, Kaariainen H, Haravuori H, Winter RM, Saamanen AM, Peltonen L, Kestila M. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum Mol Genet. 2003 Nov 1;12(21):2837-44. doi: 10.1093/hmg/ddg306. Epub 2003 Sep 2. Citation on PubMed
- Siitonen HA, Sotkasiira J, Biervliet M, Benmansour A, Capri Y, Cormier-Daire V, Crandall B, Hannula-Jouppi K, Hennekam R, Herzog D, Keymolen K, Lipsanen-Nyman M, Miny P, Plon SE, Riedl S, Sarkar A, Vargas FR, Verloes A, Wang LL, Kaariainen H, Kestila M. The mutation spectrum in RECQL4 diseases. Eur J Hum Genet. 2009 Feb;17(2):151-8. doi: 10.1038/ejhg.2008.154. Epub 2008 Aug 20. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.