

Description
Apert syndrome is a genetic disorder characterized by skeletal abnormalities. A key feature of Apert syndrome is the premature closure of the bones of the skull (craniosynostosis). This early fusion prevents the skull from growing normally and affects the shape of the head and face. In addition, a varied number of fingers and toes are fused together (syndactyly).
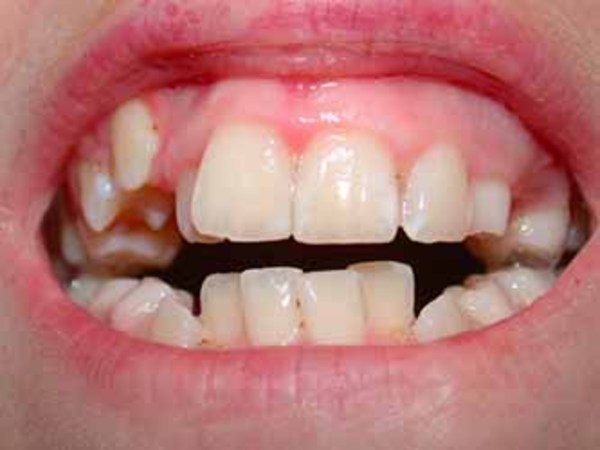
Craniosynostosis causes many of the characteristic facial features of Apert syndrome. Premature fusion of the skull bones prevents the head from growing normally, which leads to a sunken appearance in the middle of the face (midface hypoplasia), a beaked nose, a wrinkled forehead, and an opening in the roof of the mouth (a cleft palate). In individuals with Apert syndrome, an underdeveloped upper jaw can lead to dental problems, such as missing teeth, irregular tooth enamel, and crowded teeth.
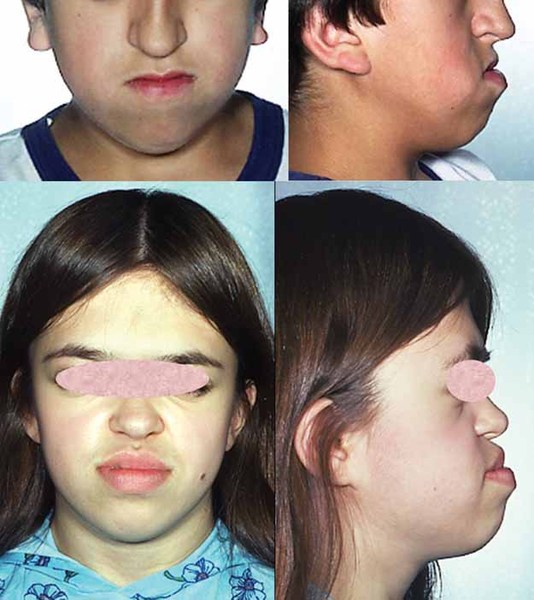

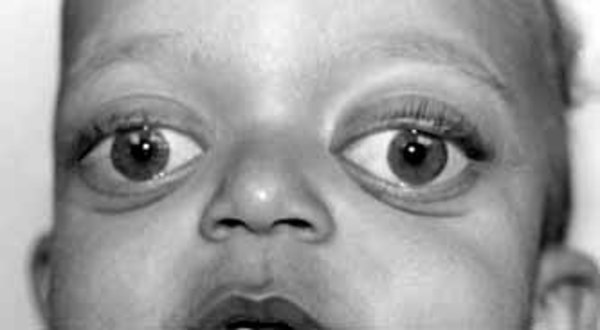
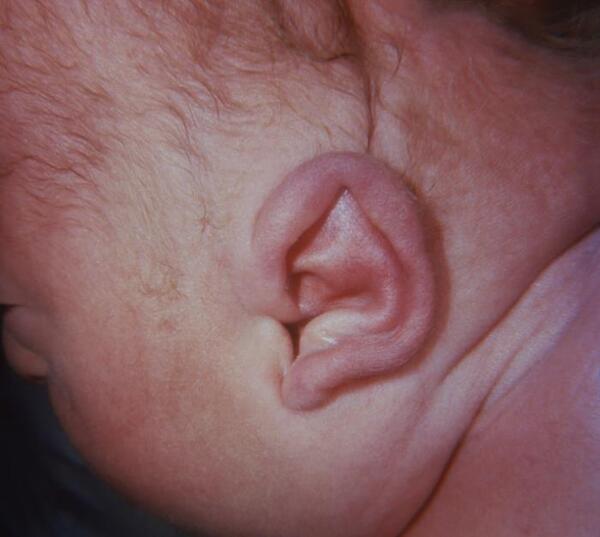
Many individuals with Apert syndrome have vision problems due to eye abnormalities, which can include bulging eyes (exophthalmos), wide-set eyes (hypertelorism), outside corners of the eyes that point downward (downslanting palpebral fissures), eyes that do not look in the same direction (strabismus), and shallow eye sockets (ocular proptosis). Some people with Apert syndrome have hearing loss or recurrent ear infections due to malformed ear structures.

Abnormal development of structures in the face and head can also cause partial blockage of the airways and lead to breathing difficulties in people with Apert syndrome. Craniosynostosis also affects development of the brain, which can disrupt intellectual development. Cognitive abilities in people with Apert syndrome range from normal to mild or moderate intellectual disability.
Individuals with Apert syndrome have syndactyly of the fingers and toes. The severity of the fusion varies, although the hands tend to be more severely affected than the feet. Most commonly, three digits on each hand and foot are fused together. In the most severe cases, all of the fingers and toes are fused. Rarely, people with Apert syndrome may have extra fingers or toes (polydactyly). Some people with Apert syndrome have abnormalities in the bones of the elbows or shoulders. These bone problems can restrict movement and impede everyday activities. In some people, abnormalities occur in both sides of the body, but in others, only one side is affected.
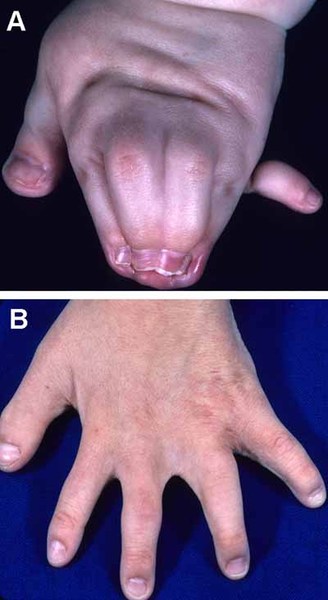
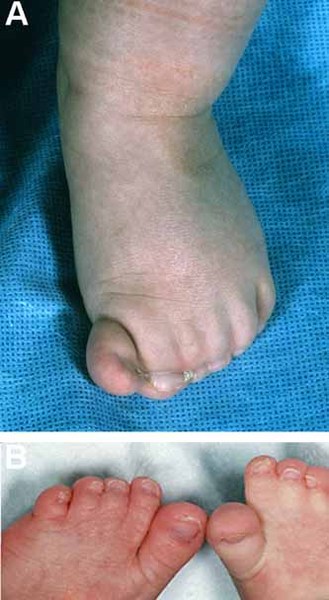
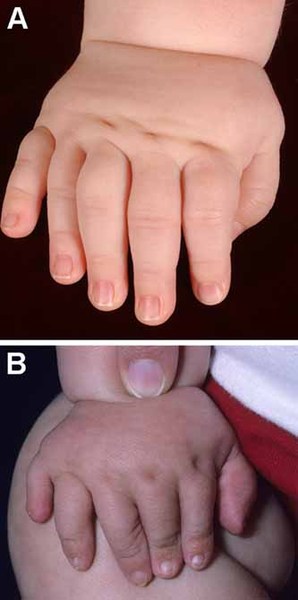
Additional signs and symptoms of Apert syndrome can include unusually heavy sweating (hyperhidrosis), oily skin with severe acne, or patches of missing hair in the eyebrows.
Frequency
Apert syndrome affects an estimated 1 in 65,000 to 88,000 newborns.
Although parents of all ages can have a child with Apert syndrome, the risk is increased in older fathers.
Causes
Mutations in a gene known as FGFR2 cause Apert syndrome. This gene provides instructions for making a protein called fibroblast growth factor receptor 2 (FGFR2). Among its multiple functions, the FGFR2 protein plays a key role in development before birth by signaling immature cells to become bone cells. A mutation in a specific part of the FGFR2 gene alters the protein, increasing its signaling. The abnormal signaling causes the cell to mature too quickly and promotes the premature fusion of bones in the skull, hands, and feet.
Inheritance
Apert syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Nearly all cases of this condition result from new (de novo) mutations in the gene that occur during the formation of reproductive cells (eggs or sperm) in an affected individual's parent or in early embryonic development. These cases occur in people with no history of the disorder in their family.

Other Names for This Condition
- Acrocephalosyndactyly
- Acrocephalosyndactyly type I
- Apert's syndrome
- Type I acrocephalosyndactyly
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Anderson PJ, Hall CM, Evans RD, Hayward RD, Jones BM. The elbow in syndromic craniosynostosis. J Craniofac Surg. 1998 May;9(3):201-6. doi: 10.1097/00001665-199805000-00002. Citation on PubMed
- Anderson PJ, Hall CM, Evans RD, Hayward RD, Jones BM. The feet in Apert's syndrome. J Pediatr Orthop. 1999 Jul-Aug;19(4):504-7. doi: 10.1097/00004694-199907000-00015. Citation on PubMed
- Anderson PJ, Hall R, Smith PJ. Finger duplication in Apert's syndrome. J Hand Surg Br. 1996 Oct;21(5):649-51. doi: 10.1016/s0266-7681(96)80151-3. Citation on PubMed
- Carinci F, Pezzetti F, Locci P, Becchetti E, Carls F, Avantaggiato A, Becchetti A, Carinci P, Baroni T, Bodo M. Apert and Crouzon syndromes: clinical findings, genes and extracellular matrix. J Craniofac Surg. 2005 May;16(3):361-8. doi: 10.1097/01.scs.0000157078.53871.11. Citation on PubMed
- Chen L, Deng CX. Roles of FGF signaling in skeletal development and human genetic diseases. Front Biosci. 2005 May 1;10:1961-76. doi: 10.2741/1671. Citation on PubMed
- David DJ, Anderson P, Flapper W, Syme-Grant J, Santoreneos S, Moore M. Apert Syndrome: Outcomes From the Australian Craniofacial Unit's Birth to Maturity Management Protocol. J Craniofac Surg. 2016 Jul;27(5):1125-34. doi: 10.1097/SCS.0000000000002709. Citation on PubMed
- Ibrahimi OA, Chiu ES, McCarthy JG, Mohammadi M. Understanding the molecular basis of Apert syndrome. Plast Reconstr Surg. 2005 Jan;115(1):264-70. Citation on PubMed
- Kutkowska-Kazmierczak A, Gos M, Obersztyn E. Craniosynostosis as a clinical and diagnostic problem: molecular pathology and genetic counseling. J Appl Genet. 2018 May;59(2):133-147. doi: 10.1007/s13353-017-0423-4. Epub 2018 Feb 1. Citation on PubMed
- Surman TL, Logan RM, Townsend GC, Anderson PJ. Oral features in Apert syndrome: a histological investigation. Orthod Craniofac Res. 2010 Feb;13(1):61-7. doi: 10.1111/j.1601-6343.2009.01478.x. Citation on PubMed
- Verma S, Draznin M. Apert syndrome. Dermatol Online J. 2005 Mar 1;11(1):15. No abstract available. Citation on PubMed
- Wenger T, Miller D, Evans K. FGFR Craniosynostosis Syndromes Overview. 1998 Oct 20 [updated 2020 Apr 30]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1455/ Citation on PubMed
- Wilkie AO, Patey SJ, Kan SH, van den Ouweland AM, Hamel BC. FGFs, their receptors, and human limb malformations: clinical and molecular correlations. Am J Med Genet. 2002 Oct 15;112(3):266-78. doi: 10.1002/ajmg.10775. Citation on PubMed
- Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995 Feb;9(2):165-72. doi: 10.1038/ng0295-165. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.