

Description
Anhidrotic ectodermal dysplasia with immune deficiency (EDA-ID) is a form of ectodermal dysplasia, which is a group of conditions characterized by abnormal development of ectodermal tissues including the skin, hair, teeth, and sweat glands. In addition, immune system function is reduced in people with EDA-ID. The signs and symptoms of EDA-ID are evident soon after birth, and due to the severity of the immune system problems, most people with this condition survive only into childhood.
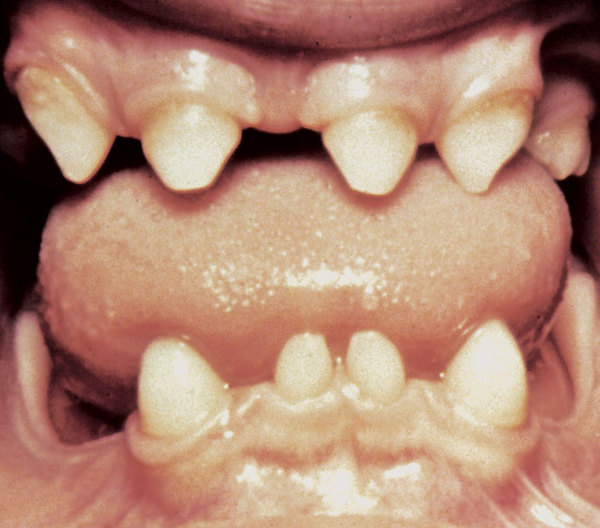
Skin abnormalities in children with EDA-ID include areas that are dry, wrinkled, or darker in color than the surrounding skin. Affected individuals tend to have sparse scalp and body hair (hypotrichosis). EDA-ID is also characterized by missing teeth (hypodontia) or teeth that are small and pointed. Most children with EDA-ID have a reduced ability to sweat (hypohidrosis) because they have fewer sweat glands than normal or their sweat glands do not function properly. An inability to sweat (anhidrosis) can lead to a dangerously high body temperature (hyperthermia), particularly in hot weather and during exercise, because the body cannot cool itself by evaporating sweat.

The immune deficiency in EDA-ID varies among individuals with this condition. Children with EDA-ID often produce abnormally low levels of proteins called antibodies or immunoglobulins. Antibodies help protect the body against infection by attaching to specific foreign particles and germs, marking them for destruction. A reduction in antibodies makes it difficult for children with this disorder to fight off infections. In EDA-ID, immune system cells called T cells and B cells have a decreased ability to recognize and respond to foreign invaders (such as bacteria, viruses, and yeast) that have sugar molecules attached to their surface (glycan antigens). Other key aspects of the immune system may also be impaired, leading to recurrent infections.
Children with EDA-ID commonly get infections in the lungs (pneumonia), ears (otitis media), sinuses (sinusitis), lymph nodes (lymphadenitis), skin, bones, and gastrointestinal tract. Approximately one quarter of individuals with EDA-ID have disorders involving abnormal inflammation, such as inflammatory bowel disease or rheumatoid arthritis.
There are two forms of EDA-ID that have similar signs and symptoms and are distinguished by the modes of inheritance: X-linked recessive or autosomal dominant.
Frequency
The prevalence of the X-linked recessive type of EDA-ID is estimated to be 1 in 250,000 individuals. Only a few cases of the autosomal dominant form have been described in the scientific literature.
Causes
Variants (also known as mutations in the IKBKG gene cause X-linked recessive EDA-ID, and variants in the NFKBIA gene cause autosomal dominant EDA-ID. The proteins produced from these two genes regulate nuclear factor-kappa-B. Nuclear factor-kappa-B is a group of related proteins (a protein complex) that binds to DNA and controls the activity of other genes, including genes that direct the body's immune responses and inflammatory reactions. It also protects cells from certain signals that would otherwise cause them to self-destruct (undergo apoptosis ).
).
The IKBKG and NFKBIA gene variants responsible for EDA-ID result in the production of proteins with altered function, which reduces activation of nuclear factor-kappa-B. These changes disrupt certain signaling pathways within immune cells, resulting in immune deficiency. It is unclear how gene variants alter the development of the skin, teeth, sweat glands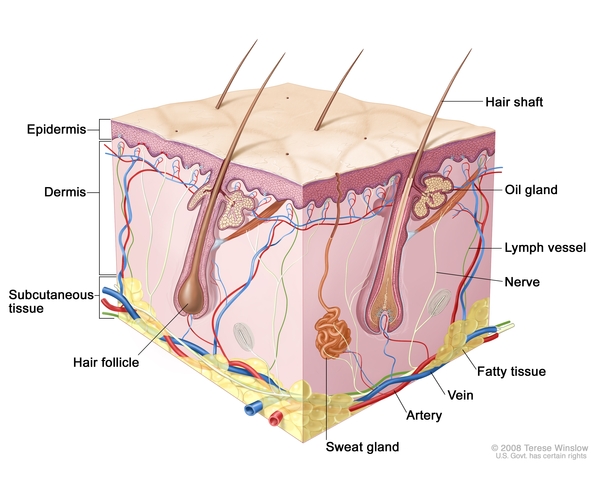 , and other tissues, although it is likely caused by abnormal nuclear factor-kappa-B signaling in other types of cells.
, and other tissues, although it is likely caused by abnormal nuclear factor-kappa-B signaling in other types of cells.
Inheritance
When EDA-ID is caused by variants in the IKBKG gene, it is inherited in an X-linked recessive pattern . The IKBKG gene is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a variant would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of the IKBKG gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
. The IKBKG gene is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a variant would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of the IKBKG gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
When EDA-ID is caused by variants in the NFKBIA gene, the condition is inherited in an autosomal dominant pattern , which means one copy of the altered gene in each cell is sufficient to cause the disorder. Most cases result from new variants in the gene and occur in people with no history of the disorder in their family.
, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Most cases result from new variants in the gene and occur in people with no history of the disorder in their family.
Other Names for This Condition
- Ectodermal dysplasia, hypohidrotic, with immune deficiency
- EDA-ID
- HED-ID
- Hyper-IgM immunodeficiency with hypohidrotic ectodermal dysplasia
- Hypohidrotic ectodermal dysplasia with immune deficiency
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Boisson B, Puel A, Picard C, Casanova JL. Human IkappaBalpha Gain of Function: a Severe and Syndromic Immunodeficiency. J Clin Immunol. 2017 Jul;37(5):397-412. doi: 10.1007/s10875-017-0400-z. Epub 2017 Jun 9. Citation on PubMed
- Hubeau M, Ngadjeua F, Puel A, Israel L, Feinberg J, Chrabieh M, Belani K, Bodemer C, Fabre I, Plebani A, Boisson-Dupuis S, Picard C, Fischer A, Israel A, Abel L, Veron M, Casanova JL, Agou F, Bustamante J. New mechanism of X-linked anhidrotic ectodermal dysplasia with immunodeficiency: impairment of ubiquitin binding despite normal folding of NEMO protein. Blood. 2011 Jul 28;118(4):926-35. doi: 10.1182/blood-2010-10-315234. Epub 2011 May 26. Citation on PubMed or Free article on PubMed Central
- Kawai T, Nishikomori R, Heike T. Diagnosis and treatment in anhidrotic ectodermal dysplasia with immunodeficiency. Allergol Int. 2012 Jun;61(2):207-17. doi: 10.2332/allergolint.12-RAI-0446. Citation on PubMed
- Mancini AJ, Lawley LP, Uzel G. X-linked ectodermal dysplasia with immunodeficiency caused by NEMO mutation: early recognition and diagnosis. Arch Dermatol. 2008 Mar;144(3):342-6. doi: 10.1001/archderm.144.3.342. Citation on PubMed
- Mooster JL, Cancrini C, Simonetti A, Rossi P, Di Matteo G, Romiti ML, Di Cesare S, Notarangelo L, Geha RS, McDonald DR. Immune deficiency caused by impaired expression of nuclear factor-kappaB essential modifier (NEMO) because of a mutation in the 5' untranslated region of the NEMO gene. J Allergy Clin Immunol. 2010 Jul;126(1):127-32.e7. doi: 10.1016/j.jaci.2010.04.026. Epub 2010 Jun 12. Citation on PubMed or Free article on PubMed Central
- Temmerman ST, Ma CA, Zhao Y, Keenan J, Aksentijevich I, Fessler M, Brown MR, Knutsen A, Shapiro R, Jain A. Defective nuclear IKKalpha function in patients with ectodermal dysplasia with immune deficiency. J Clin Invest. 2012 Jan;122(1):315-26. doi: 10.1172/JCI42534. Epub 2011 Dec 12. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.